Optimizing Tulathromycin Production: A Safer, Scalable Route For Veterinary Pharmaceutical Intermediates
The global demand for effective veterinary antibiotics continues to rise, driven by the critical need to manage respiratory diseases in livestock such as swine and cattle. Within this sector, Tulathromycin stands out as a third-generation macrolide antibiotic with exceptional efficacy against pathogens like Actinobacillus pleuropneumoniae and Pasteurella multocida. However, the commercial viability of this high-value active pharmaceutical ingredient (API) relies heavily on the efficiency and safety of its synthetic pathway. The patent CN106939029B introduces a transformative preparation method that addresses long-standing industrial bottlenecks. By replacing traditional catalytic hydrogenation with a chemically driven deprotection strategy using p-nitrobenzyl chloroformate, this technology offers a robust alternative for manufacturers seeking to optimize their production lines. This report analyzes the technical merits of this novel route, providing actionable insights for R&D directors and procurement specialists aiming to secure a reliable veterinary drug intermediate supplier.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of tulathromycin has been plagued by significant safety and cost inefficiencies inherent in earlier patented routes. The original methodology developed by Pfizer, for instance, utilized benzyl chloroformate to protect the 2'-hydroxyl group of demethylazithromycin. While chemically effective, the removal of this benzyl protecting group necessitates catalytic hydrogenation using palladium on carbon (Pd/C). This step introduces severe operational hazards, including the handling of high-pressure hydrogen gas and pyrophoric catalysts, which complicates facility safety protocols and increases insurance and maintenance costs. Furthermore, alternative routes attempting to avoid hydrogenation, such as those employing acetyl or tert-butoxycarbonyl protecting groups, have suffered from poor selectivity and stability issues. Acetyl groups often require harsh alkaline conditions for removal, which can lead to the hydrolysis of the sensitive macrolide ester ring, generating difficult-to-remove impurities and drastically reducing overall yield. These technical deficiencies create substantial barriers to entry for generic manufacturers and limit the scalability of production.
The Novel Approach
The innovative process detailed in patent CN106939029B circumvents these challenges through a strategic modification of the protecting group chemistry. By employing p-nitrobenzyl chloroformate, the synthesis creates a p-nitrobenzyloxycarbonyl intermediate that is stable during oxidation and epoxidation but can be cleaved under mild nucleophilic conditions. This eliminates the requirement for transition metal catalysts entirely. The reaction sequence proceeds through a streamlined four-step pathway: selective protection, Swern oxidation, Corey-Chaykovsky epoxidation, and a final tandem deprotection-addition step. This approach not only enhances operator safety by removing high-pressure hydrogenation units from the workflow but also simplifies downstream processing. The ability to perform deprotection and nucleophilic addition in a single pot using n-propylamine represents a significant process intensification, reducing solvent consumption and cycle time. For a reliable veterinary drug intermediate supplier, adopting this route translates directly into improved margin potential and supply chain resilience.
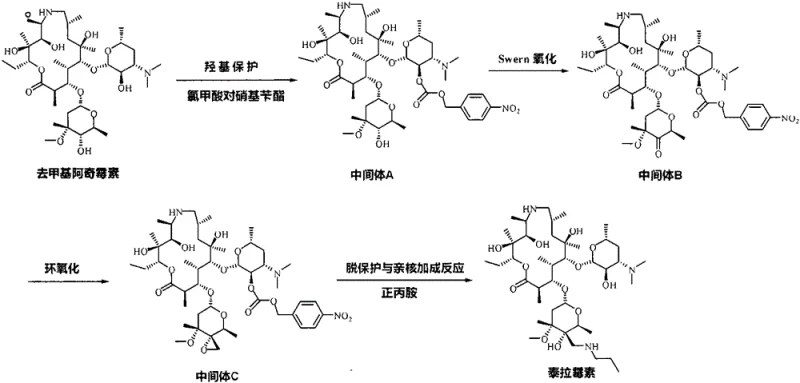
Mechanistic Insights into p-Nitrobenzyl Protection and Epoxidation Strategy
The core chemical innovation lies in the electronic properties of the p-nitrobenzyl group, which activates the carbamate linkage towards nucleophilic attack while maintaining stability against the oxidative conditions required for the 4''-hydroxyl modification. In the initial step, demethylazithromycin is reacted with p-nitrobenzyl chloroformate in solvents such as dichloromethane or tetrahydrofuran at controlled temperatures between -5°C and 0°C. The presence of the electron-withdrawing nitro group facilitates the subsequent removal of the protecting group without the need for reduction. Following protection, the 4''-hydroxyl group undergoes Swern oxidation. This reaction is conducted at cryogenic temperatures, specifically between -90°C and -60°C, using dimethyl sulfoxide (DMSO) activated by trifluoroacetic anhydride. The precise temperature control is critical here to prevent side reactions and ensure the selective formation of the ketone intermediate (Intermediate B). The patent highlights a crucial purification step where Intermediate B is isolated as a salt using acids like acetic or phosphoric acid. This salt formation acts as a powerful impurity purge, ensuring that only high-purity material enters the subsequent epoxidation stage, which is vital for controlling the stereochemistry of the final product.
The transformation of the ketone to the epoxide (Intermediate C) utilizes the Corey-Chaykovsky reaction, employing a sulfur ylide generated from trimethyl sulfonium bromide and a strong base such as sodium methoxide or potassium tert-butoxide. This step is highly sensitive to moisture and temperature, requiring reaction conditions between -80°C and -70°C to maximize yield and minimize ring-opening byproducts. The final conversion to tulathromycin is particularly elegant; the addition of n-propylamine serves a dual purpose. It acts as a nucleophile to open the epoxide ring at the 4'' position, installing the necessary amino-propyl side chain, while simultaneously attacking the p-nitrobenzyl carbamate to release the free 2'-hydroxyl group. This tandem reaction occurs at moderate temperatures of 50°C to 60°C over a period of 18 to 48 hours. By combining deprotection and functionalization, the process avoids isolating unstable intermediates, thereby reducing material loss and exposure to degradation pathways. This mechanistic efficiency is key to achieving the high purity specifications required for regulatory approval in veterinary medicine.
How to Synthesize Tulathromycin Efficiently
Implementing this synthesis requires strict adherence to the thermal profiles and stoichiometric ratios defined in the patent to ensure reproducibility and safety. The process begins with the dissolution of demethylazithromycin in a chlorinated or ethereal solvent, followed by the controlled addition of the protecting agent and a tertiary amine base. Operators must monitor the exotherm carefully during the activation of DMSO for the oxidation step to prevent thermal runaway. The subsequent epoxidation demands rigorous exclusion of water to maintain the activity of the sulfur ylide. Finally, the workup involves careful pH adjustment and solvent exchange to precipitate the final product. For detailed standard operating procedures and specific equipment requirements, please refer to the technical guide below.
- Protect the 2'-hydroxyl group of demethylazithromycin using p-nitrobenzyl chloroformate at -5 to 0°C to form Intermediate A.
- Perform Swern oxidation on the 4''-hydroxyl group of Intermediate A at -80 to -70°C using DMSO and trifluoroacetic anhydride to yield Intermediate B.
- Conduct epoxidation of the 4''-carbonyl group in Intermediate B using Corey-Chaykovsky reagent (trimethyl sulfonium bromide and base) at low temperature to generate Intermediate C.
- Execute simultaneous deprotection and nucleophilic addition by reacting Intermediate C with n-propylamine at 50-60°C to finalize tulathromycin.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers profound advantages for cost reduction in pharmaceutical manufacturing and supply chain stability. The most immediate impact is the elimination of palladium-on-carbon catalysts. Precious metal catalysts represent a significant variable cost in fine chemical production, subject to volatile market pricing and complex recovery logistics. By removing this dependency, manufacturers can stabilize their raw material costs and reduce the capital expenditure associated with hydrogenation reactors and safety systems. Furthermore, the avoidance of high-pressure hydrogen gas simplifies the regulatory compliance landscape, allowing for faster site approvals and reduced insurance premiums. The 'one-pot' nature of the final steps also contributes to substantial cost savings by minimizing solvent turnover and reducing the labor hours required for multiple isolation and drying cycles. These efficiencies compound to create a more competitive cost structure for the final API.
- Cost Reduction in Manufacturing: The removal of expensive palladium catalysts and high-pressure hydrogenation equipment drastically lowers both CAPEX and OPEX. Additionally, the use of common organic solvents and reagents like n-propylamine ensures that raw material sourcing remains stable and affordable, avoiding the supply bottlenecks often associated with specialized catalytic reagents. The streamlined workflow reduces utility consumption, particularly in terms of heating and cooling loads, further enhancing the economic viability of large-scale production runs.
- Enhanced Supply Chain Reliability: By relying on robust chemical transformations rather than sensitive catalytic processes, the risk of batch failure due to catalyst poisoning or equipment malfunction is significantly mitigated. The intermediate crystallization step provides a built-in quality gate, ensuring that only material meeting strict purity standards proceeds to the final stages. This consistency is critical for maintaining continuous supply to downstream formulation partners, reducing the likelihood of stockouts caused by out-of-specification batches. The use of widely available reagents also diversifies the supplier base, reducing dependency on single-source vendors.
- Scalability and Environmental Compliance: The process is inherently scalable, having been designed with industrial constraints in mind. The absence of heavy metal waste streams simplifies effluent treatment and reduces the environmental footprint of the manufacturing site. This aligns with increasingly stringent global environmental regulations, future-proofing the production facility against tighter emission standards. The ability to scale from pilot plant quantities to multi-ton commercial production without fundamental process changes ensures that supply can grow in lockstep with market demand for veterinary respiratory treatments.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this tulathromycin synthesis pathway. These answers are derived directly from the experimental data and beneficial effects described in the patent literature, providing clarity on process robustness and quality control measures. Understanding these details is essential for technical teams evaluating the feasibility of technology transfer.
Q: Why is the p-nitrobenzyl protection group superior to benzyl protection in tulathromycin synthesis?
A: The p-nitrobenzyl group allows for chemical deprotection using n-propylamine under mild conditions, eliminating the need for hazardous and expensive palladium-on-carbon catalytic hydrogenation required for benzyl groups. This significantly improves industrial safety and reduces heavy metal contamination risks.
Q: How does this process ensure high purity of the final veterinary antibiotic?
A: The process incorporates a salt crystallization step for Intermediate B using acids like acetic or trifluoroacetic acid. This purification stage effectively removes impurities prior to the critical epoxidation step, ensuring a cleaner reaction profile and higher quality final API.
Q: Is this synthetic route suitable for large-scale commercial manufacturing?
A: Yes, the route is designed for scalability by utilizing 'one-pot' methodologies for oxidation and deprotection steps. This reduces solvent usage, minimizes unit operations, and avoids the safety bottlenecks associated with high-pressure hydrogenation equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tulathromycin Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex synthetic routes like the p-nitrobenzyl protected tulathromycin process can be executed with precision and reliability. We maintain stringent purity specifications across all our veterinary intermediates, supported by rigorous QC labs equipped with advanced analytical instrumentation to verify identity and impurity profiles. Our commitment to quality assurance means that every batch delivered meets the exacting standards required by global regulatory bodies, providing peace of mind to our partners in the pharmaceutical industry.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. Our technical procurement team is ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized manufacturing processes can enhance your supply chain efficiency. By collaborating with NINGBO INNO PHARMCHEM, you gain access to a secure, high-quality source of veterinary drug intermediates that drives value through both technical excellence and commercial reliability. Contact us today to discuss how we can support your tulathromycin supply needs.