Advanced Gold-Catalyzed Synthesis of Etoposide for Scalable Pharmaceutical Manufacturing
Advanced Gold-Catalyzed Synthesis of Etoposide for Scalable Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for critical oncology agents, and Etoposide remains a cornerstone in the treatment of various malignancies including small cell lung cancer and leukemia. A significant technological advancement in this domain is detailed in patent CN115505017A, which discloses a novel synthesis method for Etoposide and its analogues. This innovative approach addresses long-standing challenges in stereoselective glycosylation by introducing a unique glycosyl propargyl amino carbonate donor system. Unlike conventional strategies that rely on harsh Lewis acids, this method utilizes a mild Au(I)-catalyzed protocol to couple the sugar moiety with the podophyllotoxin core. The result is a process that offers superior control over regioselectivity and stereoselectivity, ensuring the production of the therapeutically active beta-anomer with high fidelity. This technical breakthrough represents a pivotal shift towards more efficient and reliable pharmaceutical intermediates manufacturing.
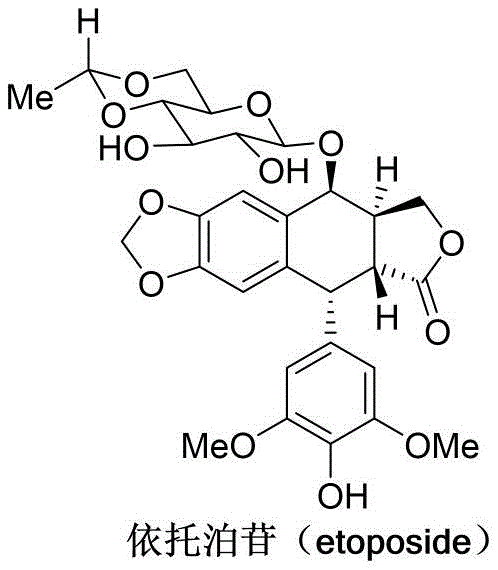
Etoposide, chemically known as 9-(4,6-O-ethylene-beta-D-glucopyranoside)-4'-demethylepipodophyllotoxin, is a complex molecule comprising a naturally sourced hexose fragment and a demethylepipodophyllotoxin aglycone. The structural complexity, particularly the acid-sensitive nature of the podophyllotoxin skeleton, has historically complicated its total synthesis and semi-synthesis. The patent highlights that the 4-hydroxyl group on the podophyllotoxin ring is situated at a benzylic position adjacent to an electron-rich aromatic system. In traditional acidic glycosylation conditions, this specific structural feature predisposes the molecule to the formation of unstable benzylic cations, leading to decomposition and loss of stereochemical integrity. By circumventing these acidic pitfalls through the use of a gold-catalyzed neutral pathway, the new method preserves the delicate stereochemistry of the drug molecule, thereby enhancing the overall quality and consistency of the final high-purity API.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Etoposide has been plagued by the limitations inherent in classical glycosylation techniques. Prior art methods, as cited in literature such as Helv. Chim. Acta and Tetrahedron Letters, predominantly employ strong Lewis acids like boron trifluoride etherate (BF3-Et2O), titanium tetrachloride (TiCl4), or silver salts to promote the coupling reaction. While these reagents are effective activators for many glycosyl donors, they create a highly acidic environment that is detrimental to the podophyllotoxin scaffold. The primary issue arises from the facile generation of a benzylic carbocation at the C4 position under these acidic conditions. This side reaction not only consumes the valuable starting material but also generates a myriad of by-products that are structurally similar to the desired product. Consequently, the resulting reaction mixture typically contains a difficult-to-separate blend of alpha and beta anomers, along with degradation products. This lack of stereocontrol necessitates extensive and costly purification processes, such as repeated column chromatography or recrystallization, which severely impact the overall yield and economic viability of the manufacturing process.
The Novel Approach
In stark contrast to the harsh conditions of the past, the novel approach described in the patent introduces a paradigm shift by utilizing a glycosyl propargyl amino carbonate donor. This donor is synthesized by reacting a protected glycosyl fragment with propargyl isocyanate under basic conditions, creating a stable intermediate that is inert to many standard conditions but highly reactive upon specific activation. The core innovation lies in the subsequent glycosylation step, where this donor reacts with a selectively protected 4'-demethylepipodophyllotoxin derivative in the presence of a gold(I) catalyst. This catalytic system operates under remarkably mild conditions, typically at temperatures ranging from 0°C to 30°C, which effectively suppresses the formation of the deleterious benzylic cation. The result is a highly selective transformation that favors the formation of the beta-glucoside linkage exclusively. This methodological improvement eliminates the need for aggressive Lewis acids, thereby simplifying the reaction workup and significantly improving the purity profile of the crude product before any purification is even attempted.
Mechanistic Insights into Au(I)-Catalyzed Glycosylation
The mechanistic elegance of this synthesis lies in the specific activation mode of the gold(I) catalyst. In the key coupling step, the Au(I) species, often generated in situ from complexes like Ph3PAuCl and a silver salt additive such as AgOTf, coordinates selectively to the alkyne functionality of the propargyl amino carbonate donor. This coordination increases the electrophilicity of the adjacent carbonyl carbon, rendering it susceptible to nucleophilic attack by the phenolic hydroxyl group of the protected podophyllotoxin acceptor. Crucially, this activation pathway does not involve the generation of free protons or strong Lewis acidic species in the bulk solution, which is why the acid-sensitive benzylic position remains intact. The transition state is stabilized by the gold-alkyne complex, guiding the nucleophile to attack from the less hindered face, thus enforcing the formation of the beta-glycosidic bond. This level of mechanistic control is essential for producing commercial scale-up of complex pharmaceutical intermediates where batch-to-batch consistency is paramount.
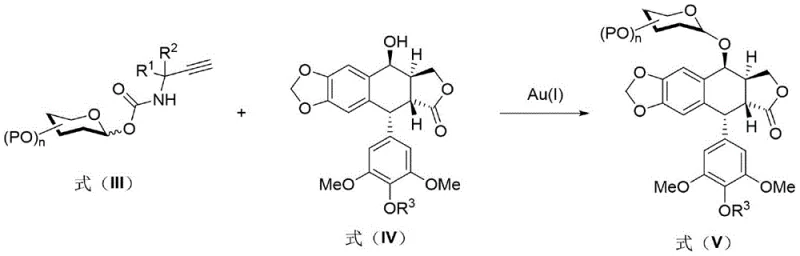
Furthermore, the choice of protecting groups plays a vital role in the success of this mechanism. The patent specifies that the 4'-phenolic hydroxyl of the aglycone must be selectively protected, for instance with a benzyloxycarbonyl (Cbz) or benzyl (Bn) group, to prevent self-reaction or polymerization. The glycosyl donor itself carries protecting groups on the sugar hydroxyls, such as benzyl or acetyl groups, which are orthogonal to the reaction conditions. The compatibility of these protecting groups with the gold catalyst ensures that the reaction proceeds cleanly without premature deprotection or migration of groups. Following the coupling, the final step involves a global deprotection strategy. Depending on the specific protecting groups chosen (e.g., benzyl ethers vs. esters), this can be achieved via catalytic hydrogenation or hydrolysis. The robustness of the intermediate beta-glucoside formed in the gold-catalyzed step ensures that it survives these final deprotection conditions intact, delivering the final Etoposide molecule with the correct stereochemistry and high chemical purity required for clinical applications.
How to Synthesize Etoposide Efficiently
The synthesis of Etoposide via this novel route involves a streamlined sequence of four distinct chemical transformations that can be executed with standard laboratory equipment and readily available reagents. The process begins with the activation of the sugar component, followed by the preparation of the aglycone, the critical gold-catalyzed coupling, and finally the removal of protecting groups. This sequence is designed to maximize yield at every stage while minimizing the formation of impurities that are difficult to remove later. For process chemists and R&D teams looking to implement this technology, the following guide outlines the critical operational parameters derived from the patent examples. It is important to note that strict control over moisture and temperature during the gold-catalyzed step is essential to maintain the activity of the catalyst and the stability of the donor. Detailed standardized synthesis steps are provided below to facilitate immediate technology transfer and process validation.
- React a protected glycosyl fragment with propargyl isocyanate under basic conditions to form a stable glycosyl propargyl amino carbonate donor.
- Selectively protect the 4'-phenolic hydroxyl group of 4'-demethylepipodophyllotoxin to obtain the protected aglycone derivative.
- Perform the glycosylation reaction between the donor and the protected aglycone using an Au(I) catalyst and a salt additive to generate the beta-glucoside with high selectivity.
- Execute global deprotection under specific conditions to remove all protecting groups and yield the final Etoposide product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this gold-catalyzed synthesis route offers substantial strategic advantages over legacy manufacturing methods. The primary benefit stems from the drastic simplification of the purification workflow. Because the reaction exhibits such high beta-selectivity, the burden on downstream processing is significantly reduced. In traditional methods, separating alpha and beta isomers often requires multiple rounds of chromatography or crystallization, which consumes vast amounts of solvents and silica gel, driving up both cost and environmental waste. By generating the correct isomer directly, this new method streamlines the production line, allowing for faster throughput and reduced consumption of consumables. This efficiency translates directly into a more competitive cost structure for the final API, making it an attractive option for generic drug manufacturers seeking cost reduction in pharmaceutical intermediates manufacturing.
- Cost Reduction in Manufacturing: The elimination of harsh Lewis acids like TiCl4 and BF3 not only improves safety but also reduces the cost associated with specialized corrosion-resistant equipment and hazardous waste disposal. Furthermore, the high yields reported in the patent examples, often exceeding 85% for the key coupling step, mean that less raw material is wasted. The ability to use milder conditions also lowers energy consumption for heating or cooling, contributing to a leaner and more cost-effective production process that enhances overall margin potential without compromising quality standards.
- Enhanced Supply Chain Reliability: The robustness of the glycosyl propargyl amino carbonate donor adds a layer of stability to the supply chain. Unlike some traditional donors that are unstable and must be prepared and used immediately, this donor can be isolated and stored, allowing for better inventory management and production scheduling. This stability reduces the risk of batch failures due to reagent degradation. Additionally, the use of gold catalysts, while precious, is required in very low loading (often around 2 mol%), and the catalyst systems are well-established in the fine chemical industry, ensuring a reliable source of supply for the necessary reagents without bottlenecking production.
- Scalability and Environmental Compliance: Scaling up reactions that involve strong Lewis acids presents significant engineering challenges due to heat generation and corrosivity. The mild, near-neutral conditions of the Au(I)-catalyzed reaction make it inherently safer and easier to scale from kilogram to multi-ton quantities. The reduction in solvent usage for purification and the avoidance of heavy metal Lewis acid waste streams align perfectly with modern green chemistry principles and increasingly stringent environmental regulations. This facilitates smoother regulatory approvals and ensures long-term operational continuity for manufacturing sites focused on sustainable pharmaceutical intermediates production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the experimental data and technical disclosures within the patent documentation. Understanding these nuances is critical for R&D teams evaluating the feasibility of adopting this route for commercial production. The focus is on resolving uncertainties regarding selectivity, catalyst recovery, and final product quality to ensure confidence in the technology transfer process.
Q: Why is the traditional glycosylation of podophyllotoxin derivatives challenging?
A: Traditional methods often utilize strong Lewis acids like BF3-Et2O or TiCl4. These acidic conditions can induce the formation of a benzylic carbocation at the C4 position of the podophyllotoxin ring due to the electron-rich nature of the benzene ring. This side reaction leads to poor stereoselectivity, difficult separation of alpha/beta isomers, and overall lower yields.
Q: What is the key advantage of using a propargyl isocyanate-derived donor?
A: The use of propargyl isocyanate allows for the formation of a glycosyl propargyl amino carbonate donor. This donor is significantly more stable than traditional trichloroacetimidates or halides. Furthermore, it enables activation under mild conditions via gold catalysis, avoiding the harsh acidic environments that degrade the sensitive podophyllotoxin core.
Q: How does the Au(I) catalyst improve the synthesis outcome?
A: The Au(I) catalyst, often used with a silver salt additive, activates the alkyne moiety of the donor specifically. This activation facilitates the nucleophilic attack by the phenolic hydroxyl group of the aglycone with exceptional beta-selectivity. This results in a highly pure beta-glucoside intermediate, drastically reducing the need for complex purification steps to remove alpha-isomers.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Etoposide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to meet the growing global demand for high-quality oncology drugs. Our technical team has thoroughly analyzed the potential of this gold-catalyzed glycosylation route and is fully equipped to translate this laboratory-scale innovation into industrial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from pilot plant to full-scale manufacturing is seamless. Our facilities are designed to handle complex organic syntheses with stringent purity specifications, supported by rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify the stereochemical integrity and purity of every batch of Etoposide we produce.
We invite global pharmaceutical partners to collaborate with us to leverage this cutting-edge technology for your supply chain. By partnering with us, you gain access to a Customized Cost-Saving Analysis that details exactly how this new route can optimize your specific procurement budget. We encourage you to contact our technical procurement team today to request specific COA data from our pilot runs and comprehensive route feasibility assessments. Let us help you secure a stable, high-quality, and cost-effective supply of Etoposide that meets the highest international regulatory standards.