Advanced Etoposide Manufacturing: High-Yield TMSOTf Catalysis for Commercial Scale
The global demand for effective antineoplastic agents continues to drive innovation in the synthesis of critical oncology drugs like Etoposide. Patent CN1376159A introduces a transformative synthetic methodology that addresses long-standing inefficiencies in the manufacturing of this vital topoisomerase II inhibitor. The disclosed technique leverages a direct condensation strategy between 4'-demethyl-epipodophyllotoxin and a specifically substituted glucopyranose derivative, catalyzed by trimethylsilyl trifluoromethane sulfonate (TMSOTf). This approach represents a significant leap forward in process chemistry, offering enhanced yields and drastically simplified isolation procedures compared to historical precedents. By optimizing the glycosylation step and refining the deprotection sequence, this patent provides a robust framework for the reliable etoposide supplier seeking to maximize output while maintaining stringent pharmaceutical standards.
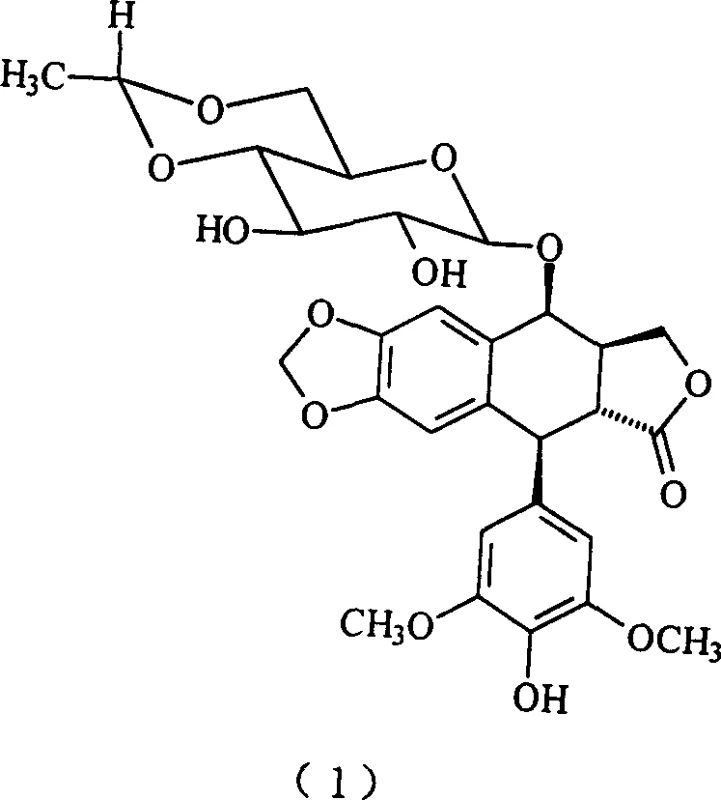
Etoposide, chemically known as 4'-demethyl-epipodophyllotoxin-4-(4,6-O-ethylidene-beta-D-glucopyranoside), has established itself as a cornerstone in the treatment of various malignancies, including acute monocytic leukemia and small cell lung cancer. The structural complexity of the molecule, particularly the stereochemical integrity of the glycosidic bond, poses significant challenges during synthesis. Traditional methods often struggle with low overall yields and the formation of difficult-to-remove impurities, such as the alpha-anomer or dimeric byproducts. The technology described in CN1376159A directly targets these pain points by introducing a novel catalytic system and a unique protecting group strategy that stabilizes the reaction intermediate, thereby ensuring high fidelity in the final product structure.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Etoposide has been plagued by multi-step sequences that inherently reduce overall efficiency and increase production costs. Early methodologies, such as those described by Kuhn et al., necessitated the protection of the 4'-phenolic hydroxyl group of the starting podophyllotoxin derivative prior to glycosylation. This additional step not only consumes reagents and time but also introduces another purification stage, leading to cumulative yield losses. Furthermore, subsequent deprotection steps often required harsh conditions that could compromise the delicate lactone ring or the glycosidic linkage. Even improved methods, such as those by Wang et al., which eliminated the phenolic protection step, still relied on boron trifluoride etherate as a catalyst, which resulted in moderate yields of approximately 54% based on the starting aglycone. These conventional routes also typically involved tedious aqueous workups, including multiple extractions and washes with acid and base, which are environmentally burdensome and operationally complex for cost reduction in pharmaceutical intermediates manufacturing.
The Novel Approach
The methodology presented in patent CN1376159A circumvents these historical bottlenecks through a streamlined two-step process that eliminates the need for phenolic protection entirely. By utilizing 2,3-di-O-dichloroacetyl-(4,6-O-ethylidene)-beta-D-glucopyranose as the glycosyl donor, the process achieves a direct coupling with 4'-demethyl-epipodophyllotoxin under mild Lewis acid catalysis. The critical innovation lies in the substitution of the traditional boron trifluoride catalyst with TMSOTf, which operates effectively at low temperatures ranging from -40°C to -60°C. This modification not only accelerates the reaction kinetics, completing the condensation within 1 to 2 hours, but also significantly enhances the stereoselectivity towards the desired beta-anomer. The resulting intermediate can be isolated through a simple filtration process using basic alumina or silica gel, bypassing the need for complex liquid-liquid extractions. This simplification of the workflow is a key driver for commercial scale-up of complex pharmaceutical intermediates, reducing both solvent consumption and processing time.
Mechanistic Insights into TMSOTf-Catalyzed Glycosylation
The superior performance of the TMSOTf-catalyzed route can be attributed to the distinct mechanistic pathway it facilitates compared to traditional boron-based Lewis acids. While boron trifluoride etherate is believed to promote glycosylation via the generation of a C4 carbocation intermediate on the sugar moiety, which can lead to scrambling of stereochemistry and the formation of the undesired alpha-glucoside, TMSOTf operates through a different activation mode. It is hypothesized that TMSOTf reacts directly with the free hydroxyl groups of the glucopyranose donor to form a highly reactive silylated leaving group. This activation promotes a concerted nucleophilic attack by the 4'-hydroxyl of the podophyllotoxin aglycone, preserving the stereochemical configuration of the starting sugar. This mechanism ensures that the beta-configuration is retained throughout the coupling process, minimizing the formation of the alpha-anomer impurity which is notoriously difficult to separate from the final active pharmaceutical ingredient.
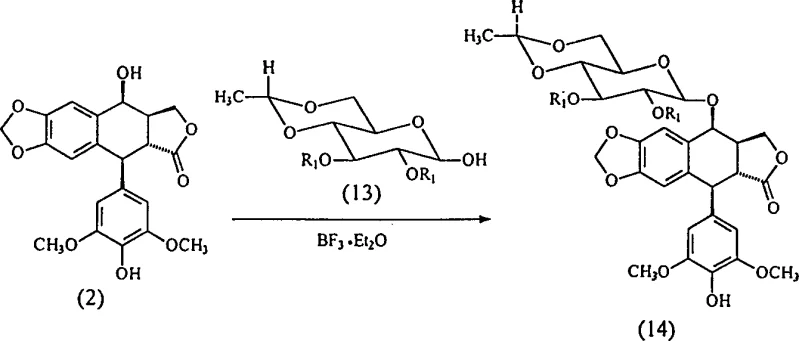
Furthermore, the choice of the dichloroacetyl protecting group on the sugar donor plays a pivotal role in the stability and reactivity of the intermediate. Unlike the chloroacetyl groups used in previous art, the dichloroacetyl moiety provides enhanced electron-withdrawing characteristics that stabilize the glycosidic bond during the reaction while remaining susceptible to mild transesterification conditions in the final step. The patent data indicates that this specific combination of catalyst and protecting group results in an intermediate yield of 80%, a substantial improvement over the 60% yield observed when using boron trifluoride under similar conditions. This mechanistic efficiency translates directly into a cleaner reaction profile, reducing the burden on downstream purification processes and ensuring that the final Etoposide product is substantially free of dimeric impurities and optical isomers, meeting the rigorous purity specifications required for oncology therapeutics.
How to Synthesize Etoposide Efficiently
The synthesis protocol outlined in the patent offers a practical and scalable route for producing high-purity Etoposide suitable for clinical applications. The process begins with the preparation of the glycosyl donor, followed by the critical low-temperature coupling reaction and a final deprotection step. Each stage is designed to maximize yield and minimize impurity formation, leveraging the unique reactivity of the TMSOTf catalyst system. Operators must maintain strict temperature control during the condensation phase to ensure optimal stereoselectivity, while the final crystallization step guarantees the removal of residual solvents and trace byproducts. For a detailed breakdown of the specific reagent quantities, temperature profiles, and workup procedures, please refer to the standardized synthesis guide below.
- Condense 4'-demethyl-epipodophyllotoxin with 2,3-di-O-dichloroacetyl-glucopyranose using TMSOTf catalyst at -40°C to -60°C.
- Isolate the intermediate glucopyranoside (14) via filtration through basic alumina or silica gel columns.
- Perform alcoholysis using zinc acetate dihydrate in methanol to remove protecting groups and crystallize final Etoposide.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of the synthesis method described in CN1376159A offers compelling economic and operational benefits. The transition from multi-step, protection-heavy routes to this direct condensation strategy fundamentally alters the cost structure of Etoposide production. By eliminating the need for phenolic protection and deprotection steps, the process reduces the total number of unit operations, which directly correlates to lower labor costs, reduced equipment occupancy time, and decreased utility consumption. The significant improvement in overall yield, rising from historical benchmarks of roughly 54% to nearly 68% based on the starting aglycone, means that less raw material is required to produce the same amount of finished API. This efficiency gain is a powerful lever for cost reduction in pharmaceutical intermediates manufacturing, allowing for more competitive pricing in a crowded generic market.
- Cost Reduction in Manufacturing: The elimination of expensive protecting group reagents and the reduction in solvent usage due to simplified workup procedures contribute to a leaner manufacturing process. The higher yield per batch means that fixed costs are amortized over a larger output of saleable product, significantly improving the margin profile. Additionally, the use of TMSOTf, while a specialized reagent, is offset by the drastic reduction in reaction time and the avoidance of yield-losing purification steps associated with older technologies.
- Enhanced Supply Chain Reliability: The robustness of the TMSOTf-catalyzed reaction makes the supply chain less vulnerable to variability. The simplified isolation method, which relies on filtration rather than complex extractions, reduces the risk of batch failures due to emulsion formation or phase separation issues. This reliability ensures consistent lead times for high-purity pharmaceutical intermediates, allowing downstream formulation partners to plan their production schedules with greater confidence. The ability to source key starting materials like 4'-demethyl-epipodophyllotoxin and convert them efficiently adds resilience to the overall supply network.
- Scalability and Environmental Compliance: From an environmental perspective, the reduction in aqueous waste streams is a major advantage. Traditional methods generate significant volumes of acidic and basic wastewater during the extraction phases, requiring costly treatment before discharge. The new method's reliance on column filtration and recrystallization minimizes liquid waste generation, aligning with modern green chemistry principles. This ease of waste management facilitates smoother regulatory approvals and supports the commercial scale-up of complex pharmaceutical intermediates without the need for massive expansions in wastewater treatment infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis route. These answers are derived directly from the experimental data and claims within patent CN1376159A, providing clarity on the feasibility and advantages of the technology for potential partners and licensees.
Q: What is the primary advantage of using TMSOTf over BF3·Et2O in Etoposide synthesis?
A: According to patent CN1376159A, TMSOTf provides significantly higher yields (80% for the intermediate vs 60% with BF3) and reduces reaction time to under 2 hours, while minimizing the formation of unwanted alpha-anomers.
Q: How does this method improve impurity control compared to conventional routes?
A: The novel isolation procedure using basic alumina filtration avoids the extensive aqueous workups required in older methods, substantially reducing the formation of 4'-demethyl-4-epipodophyllotoxin dimers and ensuring the final product is substantially free of alpha-glucoside forms.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the process eliminates the need for protecting group manipulation on the phenolic hydroxyl of the starting material and utilizes robust low-temperature conditions, making it highly scalable for industrial manufacturing of pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Etoposide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and reliable synthesis routes for life-saving oncology medications. Our technical team has extensively analyzed the advancements presented in CN1376159A and integrated similar high-efficiency protocols into our own manufacturing capabilities. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volumetric demands of global pharmaceutical clients. Our facilities are equipped with state-of-the-art cryogenic reactors capable of maintaining the precise -40°C to -60°C temperatures required for the TMSOTf-catalyzed step, guaranteeing the stereochemical purity of every batch. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that our Etoposide and related intermediates are substantially free of dimers and alpha-anomers, meeting the highest international pharmacopoeia standards.
We invite you to collaborate with us to leverage these technological advancements for your supply chain. Our experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our optimized processes can reduce your total cost of ownership. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us partner with you to secure a stable, high-quality supply of this essential anticancer agent.