Advanced FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-up
Advanced FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-up
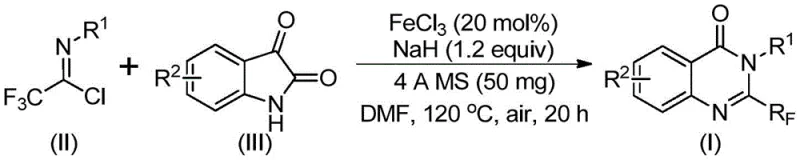
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing nitrogen-containing heterocycles, particularly quinazolinones, which serve as critical scaffolds in numerous bioactive molecules ranging from anti-cancer agents to antifungal drugs. A significant breakthrough in this domain is detailed in Chinese Patent CN111675662B, published on October 22, 2021, which discloses a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. This technology addresses long-standing challenges in heterocyclic chemistry by utilizing a cost-effective iron-catalyzed cyclization strategy that operates under relatively mild conditions compared to traditional noble metal catalysis. For R&D directors and procurement specialists alike, this patent represents a pivotal shift towards more sustainable and economically viable manufacturing processes for high-value pharmaceutical intermediates. The introduction of the trifluoromethyl group is particularly strategic, as it significantly enhances the metabolic stability, lipophilicity, and bioavailability of the final drug candidates, making this synthetic route indispensable for modern medicinal chemistry programs targeting complex disease states.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinone derivatives bearing trifluoromethyl functionalities has been plagued by substantial technical and economic hurdles that hinder efficient commercial production. Conventional literature methods typically rely on the cyclization of synthons containing trifluoromethyl groups, such as trifluoroacetic anhydride or ethyl trifluoroacetate, with substrates like anthranilamide or isatoic anhydride. These traditional pathways are frequently characterized by severe reaction conditions that demand rigorous exclusion of moisture and oxygen, often requiring expensive inert gas protections and specialized equipment. Furthermore, the starting materials employed in these legacy processes, particularly activated trifluoroacetylating agents, are not only costly but also pose significant safety hazards due to their corrosive nature and high reactivity. The narrow substrate scope of these older methods often results in poor yields when electron-withdrawing or sterically hindered groups are present, leading to extensive purification challenges and increased waste generation. Consequently, scaling these processes to an industrial level often becomes prohibitively expensive, creating bottlenecks in the supply chain for critical API intermediates.
The Novel Approach
In stark contrast to these cumbersome legacy techniques, the methodology described in patent CN111675662B introduces a streamlined and atom-economical route that leverages readily available isatin derivatives and trifluoroethylimidoyl chloride as key building blocks. This innovative approach utilizes ferric chloride (FeCl3) as a cheap and abundant Lewis acid catalyst, effectively replacing expensive transition metals like palladium or rhodium that are common in cross-coupling reactions. The reaction proceeds through a tandem sequence involving alkali-promoted carbon-nitrogen bond formation followed by an iron-catalyzed decarbonylation and cyclization, ultimately delivering the target 2-trifluoromethyl quinazolinone in high yields. By operating in common polar aprotic solvents like DMF and tolerating exposure to air, this new protocol drastically simplifies the operational complexity, removing the need for stringent anaerobic conditions. This shift not only reduces the capital expenditure required for reactor setup but also significantly lowers the barrier to entry for manufacturing these complex heterocycles, positioning it as a superior choice for reliable pharmaceutical intermediate supplier networks seeking to optimize their production portfolios.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
From a mechanistic perspective, the success of this transformation lies in the unique ability of the iron catalyst to facilitate a cascade of bond-forming and bond-breaking events with high precision. The reaction initiates with the deprotonation of the isatin nitrogen by sodium hydride, generating a nucleophilic species that attacks the electrophilic carbon of the trifluoroethylimidoyl chloride. This initial step forms a transient trifluoroacetamidine intermediate, which serves as the precursor for the subsequent cyclization. The ferric chloride then coordinates with the carbonyl oxygen and the imine nitrogen, activating the system for an intramolecular nucleophilic attack that closes the six-membered quinazolinone ring. Crucially, the iron center promotes a decarbonylation step that expels carbon monoxide, driving the equilibrium towards the formation of the stable aromatic quinazolinone core. This mechanism is remarkably robust, exhibiting excellent tolerance for a wide array of functional groups including halogens, alkyl chains, and nitro groups, which allows for the late-stage diversification of the molecular scaffold. Such mechanistic elegance ensures that impurities arising from side reactions are minimized, thereby simplifying the downstream purification process and enhancing the overall purity profile of the final product.
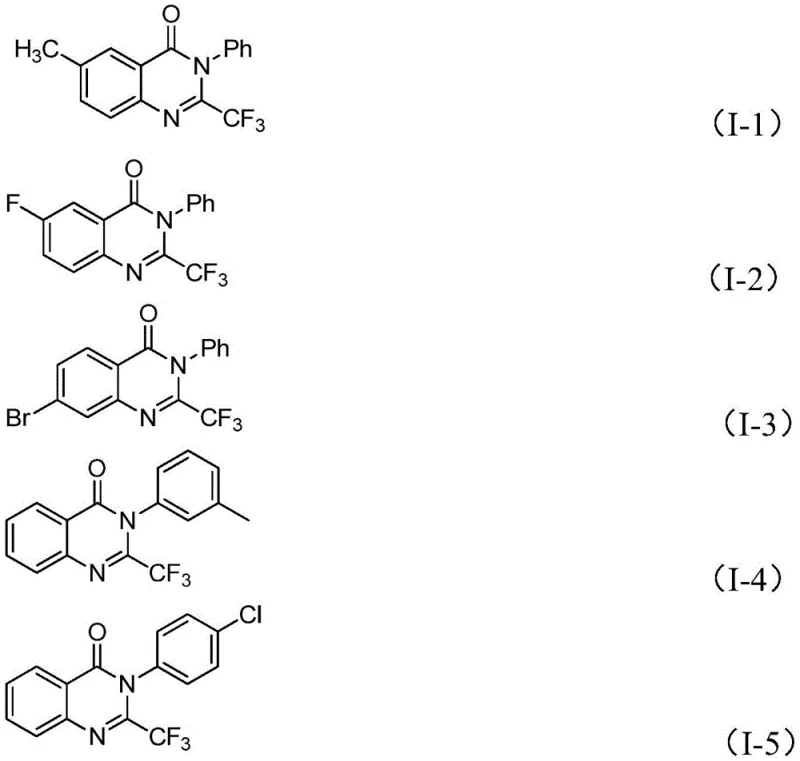
Furthermore, the structural integrity of the trifluoromethyl group is meticulously preserved throughout this catalytic cycle, which is vital for maintaining the desired pharmacokinetic properties of the end molecule. The use of 4A molecular sieves in the reaction mixture plays a critical role in scavenging trace amounts of water that could otherwise hydrolyze the sensitive imidoyl chloride starting material or deactivate the Lewis acid catalyst. This attention to detail in the reaction design underscores the practicality of the method for industrial applications where reproducibility is paramount. The ability to synthesize diverse analogues, such as those depicted in the patent examples with varying substituents at the R1 and R2 positions, demonstrates the versatility of this catalytic system. For process chemists, understanding these mechanistic nuances is essential for troubleshooting and optimizing the reaction parameters during scale-up, ensuring that the high yields observed on the gram scale can be faithfully translated to multi-kilogram production batches without compromising quality.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The practical implementation of this synthesis is designed to be straightforward, minimizing the need for specialized handling while maximizing output. The protocol involves mixing the catalyst, base, and drying agent with the substrates in a solvent like DMF, followed by a two-stage heating profile that first initiates the coupling at a lower temperature before driving the cyclization at elevated heat. This controlled thermal ramping is crucial for managing the exothermicity of the initial bond formation and ensuring complete conversion during the cyclization phase. Detailed standardized synthesis steps see the guide below.
- Combine ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in anhydrous DMF.
- Stir the mixture at 40°C for 10 hours to initiate the reaction, then increase temperature to 120°C and continue stirring under air for 20 hours.
- Upon completion, filter the reaction mixture, adsorb onto silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iron-catalyzed methodology offers transformative benefits that directly impact the bottom line and operational resilience. The primary advantage lies in the drastic reduction of raw material costs, as the process replaces expensive noble metal catalysts and hazardous acylating agents with commodity chemicals like ferric chloride and isatin. This substitution not only lowers the direct cost of goods sold but also mitigates the risks associated with the volatility of precious metal markets. Additionally, the simplified workup procedure, which relies on standard filtration and chromatography rather than complex extraction or distillation sequences, reduces the consumption of solvents and energy, contributing to a leaner and more environmentally compliant manufacturing footprint. These factors collectively enhance the economic viability of producing high-purity pharmaceutical intermediates, making it an attractive option for companies aiming to reduce lead time for high-purity intermediates while maintaining strict quality standards.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts such as palladium or rhodium removes the necessity for expensive metal scavenging steps, which are often required to meet stringent regulatory limits on residual metals in APIs. By utilizing iron, a base metal that is orders of magnitude cheaper and more abundant, the process inherently lowers the catalyst cost per kilogram of product. Furthermore, the use of commercially available isatin derivatives avoids the multi-step synthesis of specialized starting materials, streamlining the supply chain and reducing inventory holding costs. The high atom economy of the reaction ensures that a greater proportion of the input mass is converted into the desired product, minimizing waste disposal fees and maximizing resource efficiency.
- Enhanced Supply Chain Reliability: The reliance on widely sourced commodities like ferric chloride and DMF ensures that the production of these intermediates is not vulnerable to the supply disruptions that often plague specialized reagents. Since the reaction tolerates air and uses robust reagents, the logistical requirements for transportation and storage are less stringent, allowing for broader sourcing options and reduced lead times. This reliability is critical for maintaining continuous production schedules in the fast-paced pharmaceutical sector, where delays in intermediate supply can halt entire drug development programs. The ability to source raw materials from multiple global suppliers further strengthens the supply chain against geopolitical or regional instabilities.
- Scalability and Environmental Compliance: The protocol's demonstration of gram-level synthesis with simple post-treatment indicates a clear path to commercial scale-up without the need for exotic high-pressure or cryogenic equipment. The reaction conditions are compatible with standard stainless steel reactors, facilitating a seamless transition from pilot plant to full-scale manufacturing. Moreover, the reduced generation of hazardous waste and the avoidance of toxic heavy metals align with increasingly strict environmental regulations, reducing the compliance burden on manufacturing facilities. This sustainability profile not only future-proofs the production process but also enhances the corporate social responsibility standing of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, derived directly from the patent specifications and experimental data. These insights are intended to clarify the operational feasibility and strategic value of adopting this route for your specific manufacturing needs. Understanding these details is crucial for making informed decisions about process integration and supplier selection.
Q: What are the key advantages of this FeCl3-catalyzed method over traditional synthesis?
A: This method utilizes inexpensive iron catalysts and readily available isatin substrates, avoiding the harsh conditions and expensive trifluoroacetic anhydride reagents required by conventional routes, resulting in higher yields and better functional group tolerance.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly demonstrates gram-level synthesis with simple post-treatment procedures like filtration and column chromatography, indicating strong potential for commercial scale-up due to the use of robust, non-sensitive reagents.
Q: What is the substrate scope for the R1 and R2 positions?
A: The reaction exhibits excellent functional group tolerance, accommodating various substituents including alkyl, halogen (F, Cl, Br, I), methoxy, and nitro groups at ortho-, meta-, or para-positions on the aryl rings without significant loss in yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing advanced synthetic technologies to drive innovation in drug discovery and development. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the laboratory bench to the global market. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone delivered meets the highest international standards. We are committed to leveraging our technical expertise to optimize this iron-catalyzed process, delivering cost-effective solutions that accelerate your time to market.
We invite you to engage with our technical procurement team to discuss how this novel synthesis route can be tailored to your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits of switching to this methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate our capability as your trusted partner in the synthesis of complex pharmaceutical intermediates.