Advanced Synthesis of Sugammadex Sodium Diphenyl Phosphate Impurity for Global Pharma Supply Chains
Advanced Synthesis of Sugammadex Sodium Diphenyl Phosphate Impurity for Global Pharma Supply Chains
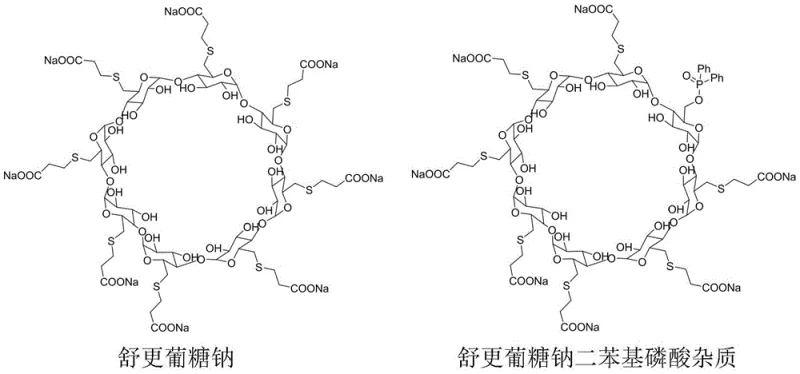
The pharmaceutical industry continuously demands higher standards for impurity profiling, particularly for complex macrocyclic APIs like Sugammadex Sodium. Patent CN111961145A introduces a groundbreaking methodology for the directed synthesis of a specific diphenyl phosphate derivative impurity found in the gamma-ICD intermediate stage. This innovation addresses a critical gap in quality control by providing a reliable pathway to generate authenticated reference standards, which are essential for regulatory compliance and batch release testing. By establishing a controlled synthetic route, manufacturers can now accurately quantify this specific process-related impurity, ensuring the highest safety profiles for the final muscle relaxant reversal agent. This technical advancement underscores the importance of proactive impurity management in modern drug development pipelines.
In the context of global supply chains, the ability to synthesize specific impurities on demand transforms a potential bottleneck into a manageable quality assurance step. Traditionally, the presence of such impurities was often detected retrospectively, leading to costly batch rejections or delays in regulatory filings. The availability of a robust synthesis method for this diphenyl phosphate derivative empowers quality control laboratories to validate their analytical methods with precision. For procurement and supply chain leaders, this translates to reduced risk of supply disruption caused by unexpected impurity spikes, thereby securing the continuity of API production for critical care medications used in anesthesia.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the identification and quantification of trace impurities in cyclodextrin-based drugs relied heavily on isolating these substances from crude reaction mixtures, a process that is notoriously inefficient and unpredictable. Conventional approaches often suffered from low recovery rates and inconsistent purity levels, making it difficult to establish definitive structural confirmation or create stable reference materials. Without a dedicated synthetic route, the diphenyl phosphate impurity would only appear sporadically during the main API synthesis, dependent on variable reaction conditions and reagent grades. This lack of control meant that pharmaceutical companies faced significant challenges in validating their HPLC methods, as they lacked authentic standards to calibrate their instruments against. Consequently, the risk of false negatives or inaccurate quantification remained a persistent threat to product quality and patient safety.
The Novel Approach
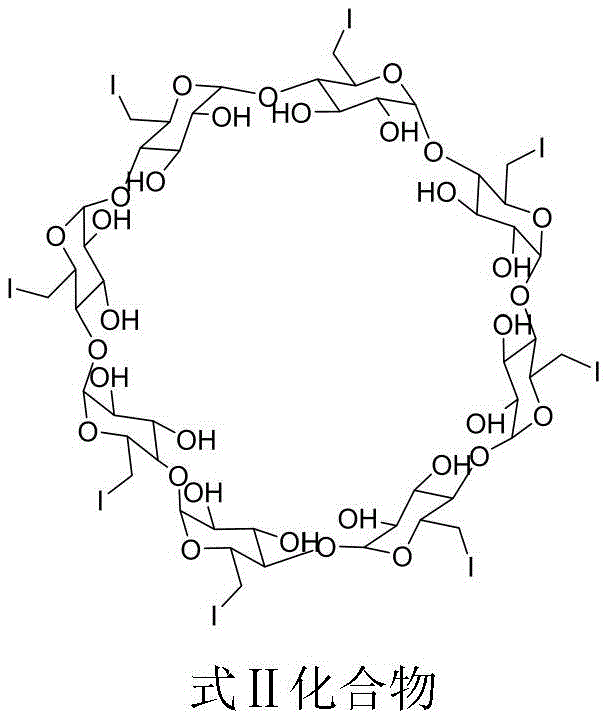
The novel approach detailed in the patent utilizes a targeted nucleophilic substitution strategy to construct the impurity molecule with high fidelity and reproducibility. By reacting 6-fully-deoxy-6-fully-iodo-gamma-cyclodextrin (gamma-ICD) with diphenyl phosphoric acid under controlled basic conditions, the process ensures the specific formation of the phosphate ester linkage at the desired position. This method eliminates the randomness associated with incidental impurity formation, allowing for the production of the target compound in substantial quantities with defined characteristics. The use of specific solvents and bases optimizes the reaction kinetics, resulting in a crude product that is amenable to straightforward purification. This shift from passive detection to active synthesis represents a paradigm shift in how complex pharmaceutical impurities are managed, offering a reliable pharmaceutical intermediate supplier pathway for critical reference materials.
Mechanistic Insights into Base-Catalyzed Phosphorylation
The core of this synthesis lies in a base-catalyzed nucleophilic substitution where the hydroxyl groups of the cyclodextrin scaffold are selectively modified. The reaction initiates with the deprotonation of diphenyl phosphoric acid by a strong base, such as cesium carbonate or sodium hydride, generating a reactive phosphate anion. This anion then attacks the electrophilic centers on the gamma-ICD intermediate, displacing the iodine atoms or reacting with available hydroxyls depending on the specific mechanistic pathway intended for this derivative. The choice of solvent, typically polar aprotic media like N,N-dimethylformamide (DMF) or dimethyl sulfoxide (DMSO), is crucial for stabilizing the ionic intermediates and facilitating the dissolution of the bulky cyclodextrin substrate. Maintaining the reaction temperature between 60-65°C provides the necessary activation energy to drive the substitution to completion without degrading the sensitive macrocyclic structure.
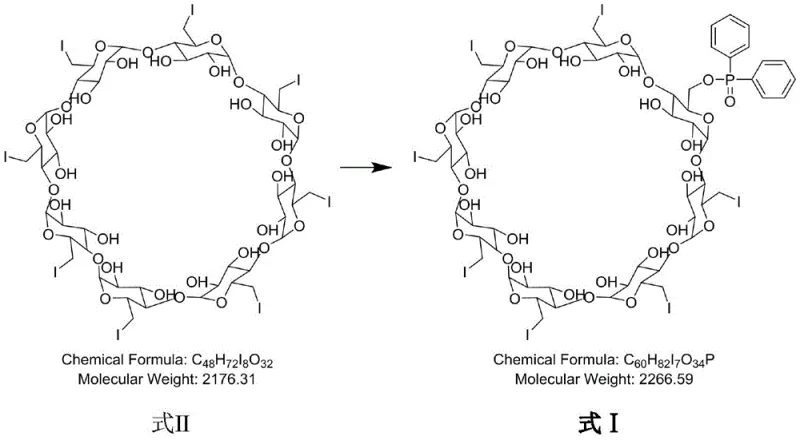
Furthermore, the control of impurity profiles is inherently built into the reaction design through the precise stoichiometry and addition order of reagents. By adding the base at low temperatures (below 10°C) prior to introducing the cyclodextrin derivative, the system minimizes side reactions such as elimination or degradation of the phosphoric acid reagent. The subsequent extended reaction time of approximately 18 hours ensures that the equilibrium favors the formation of the desired diphenyl phosphate derivative. Post-reaction processing involves a simple aqueous workup where the product precipitates or can be extracted, followed by preparative chromatography to achieve purity levels exceeding 99%. This rigorous control over the chemical environment ensures that the resulting reference standard is free from confounding byproducts, making it ideal for sensitive analytical applications in regulated environments.
How to Synthesize Sugammadex Sodium Impurity Efficiently
The synthesis protocol outlined in the patent provides a clear, step-by-step framework for producing this critical impurity standard with high consistency. It begins with the preparation of the phosphate salt in situ, followed by the controlled addition of the cyclodextrin intermediate, and concludes with a robust purification sequence. This structured approach minimizes operator variability and ensures that the critical quality attributes of the final material are met consistently. For laboratory managers and process chemists, adopting this standardized method reduces the time spent on method development and troubleshooting. The detailed synthesis steps provided in the patent serve as a foundational guide for scaling this process from milligram-scale analytical standard preparation to larger batches required for extensive stability studies.
- Dissolve diphenyl phosphoric acid in an organic solvent like DMF or DMSO and add a base such as cesium carbonate under inert gas protection at temperatures below 10°C.
- Add 6-fully-deoxy-6-fully-iodo-gamma-cyclodextrin (gamma-ICD) to the reaction mixture and maintain the temperature between 60-65°C for approximately 18 hours.
- Perform post-treatment by cooling, adding water, filtering the crude product, and purifying via preparative chromatography to achieve high purity standards.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the implementation of this patented synthesis method offers tangible strategic benefits beyond mere technical compliance. By securing a reliable internal or external source for this specific impurity standard, organizations can decouple their quality control timelines from the uncertainties of natural impurity accumulation. This independence significantly reduces the lead time for high-purity reference standards, ensuring that batch release testing proceeds without interruption. Moreover, the use of commercially available starting materials and common solvents means that the cost of goods for producing this standard is predictable and manageable, avoiding the premium pricing often associated with custom-synthesized rare impurities.
- Cost Reduction in Manufacturing: The ability to synthesize this impurity in-house or source it from a specialized provider eliminates the need for expensive and time-consuming isolation from bulk API batches. This direct synthesis route avoids the massive material loss associated with purification from complex reaction mixtures, leading to substantial cost savings in the long run. Furthermore, the mild reaction conditions reduce energy consumption and equipment wear, contributing to a more efficient overall operational expenditure for quality control departments.
- Enhanced Supply Chain Reliability: Dependence on unpredictable impurity formation poses a risk to supply continuity; this method mitigates that risk by providing a deterministic production pathway. With a stable supply of the reference standard, pharmaceutical companies can maintain consistent quality metrics across different production sites and batches. This reliability is crucial for maintaining regulatory standing and avoiding costly recalls or audit findings related to inadequate impurity monitoring systems.
- Scalability and Environmental Compliance: The process utilizes standard organic solvents and bases that are well-understood in terms of waste treatment and environmental impact. The scalability of the reaction from small laboratory flasks to larger reactors is straightforward due to the absence of exotic catalysts or extreme pressure requirements. This ease of scale-up ensures that as production volumes of the parent API increase, the supply of the necessary impurity standard can grow in tandem without requiring significant capital investment in new infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of this diphenyl phosphate derivative. Understanding these details helps stakeholders appreciate the value proposition of this patented technology in the broader context of pharmaceutical manufacturing. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals.
Q: What is the significance of the diphenyl phosphate impurity in Sugammadex Sodium production?
A: This impurity is a critical process-related substance that must be monitored to ensure the safety and efficacy of the final API. Having a certified reference standard allows for accurate quantification during quality control.
Q: How does the patented method improve upon traditional impurity synthesis?
A: The method described in CN111961145A offers mild reaction conditions, high stability, and superior yield compared to uncontrolled formation during API synthesis, ensuring a reliable supply of the reference standard.
Q: Can this synthesis process be scaled for commercial reference material production?
A: Yes, the use of common organic solvents like DMF and standard bases like cesium carbonate, along with moderate temperatures, makes the process highly scalable for industrial manufacturing of reference standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sugammadex Sodium Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality impurity standards play in the successful commercialization of complex pharmaceuticals like Sugammadex Sodium. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need milligrams for analysis or kilograms for process validation, we can deliver. We operate with stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of reference material meets the highest international standards. Our commitment to technical excellence means we can replicate the precise conditions described in patent CN111961145A to provide you with authenticated materials that support your regulatory filings.
We invite you to collaborate with us to optimize your supply chain for Sugammadex Sodium intermediates and related impurities. By requesting a Customized Cost-Saving Analysis, you can discover how our manufacturing efficiencies can translate into better margins for your organization. We encourage you to contact our technical procurement team today to索取 specific COA data and route feasibility assessments tailored to your project's unique requirements. Let us be your partner in navigating the complexities of pharmaceutical impurity management with confidence and precision.