Advanced Synthesis of 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenyl Pyrimidine-2-amine Derivatives for Commercial Scale-up
Introduction to Next-Generation EGFR Inhibitor Synthesis
The pharmaceutical landscape for non-small cell lung cancer (NSCLC) treatment is rapidly evolving, driven by the critical need for third-generation EGFR inhibitors that overcome drug resistance mechanisms such as the T790M mutation. Patent CN113773304A discloses a groundbreaking preparation method for 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenyl pyrimidine-2-amine derivatives, specifically targeting the synthesis of compounds with the general formula (IV). This technology represents a significant leap forward in process chemistry, addressing the longstanding challenges of cost, safety, and scalability associated with producing these vital pharmaceutical intermediates. By re-engineering the synthetic pathway from the ground up, this method offers a robust alternative to legacy processes that rely on expensive, hard-to-source starting materials.
For R&D directors and procurement managers alike, the implications of this patent are profound. The disclosed route not only enhances the purity profile of the final active ingredient but also streamlines the manufacturing workflow by eliminating cumbersome purification steps. As a reliable pharmaceutical intermediates supplier, understanding these technical nuances is essential for securing a stable supply chain. The method leverages widely available raw materials like indole and 2,4-dichloropyrimidine, bypassing the bottlenecks of previous generations. This strategic shift ensures that production can be scaled from kilogram to multi-ton quantities without compromising on quality or timeline, directly supporting the global demand for effective anti-tumor therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those disclosed in WO2016054987 by Jiangsu Hawson, have historically relied on 3-(2-chloropyrimidin-4-yl)-1-cyclopropyl-1H-indole as a key starting material. However, this specific intermediate is notoriously difficult to procure on a large industrial scale, creating a significant supply chain vulnerability. Furthermore, academic precedents like the method in J. Org. Lett. 2008 utilize cyclopropylboronic acid in large excess, coupled with toxic catalysts such as DMAP and copper acetate in high-boiling solvents like toluene at temperatures reaching 95°C. These conditions not only inflate the cost reduction in pharmaceutical intermediates manufacturing efforts but also impose heavy environmental burdens due to solvent recovery and toxic waste disposal.
The reliance on silica gel column chromatography for purification in older methods further exacerbates operational inefficiencies. This technique is labor-intensive, generates substantial solid waste, and is practically impossible to implement in large-scale commercial reactors. The combination of high energy consumption, toxic reagents, and complex downstream processing renders these conventional routes economically unviable for mass production. Consequently, manufacturers face prolonged lead times and inconsistent batch quality, which are unacceptable in the highly regulated context of oncology drug production. These structural deficiencies in the legacy supply chain necessitate a fundamental rethinking of the synthetic strategy.
The Novel Approach
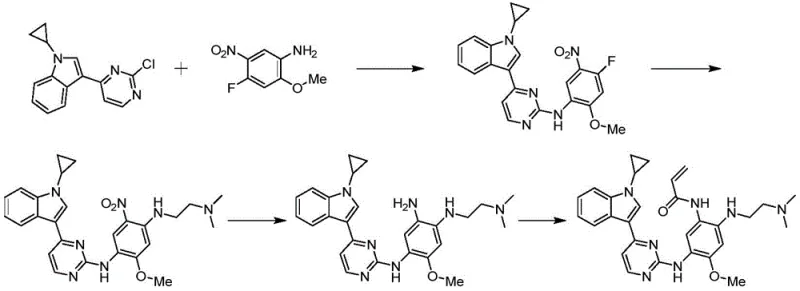
The innovative process outlined in CN113773304A fundamentally重构 s the synthetic logic by constructing the core scaffold from inexpensive, commodity chemicals. Instead of purchasing the pre-functionalized indole, the new method synthesizes it in situ via a Grignard reaction between indole and 2,4-dichloropyrimidine. This is followed by a highly optimized copper-catalyzed coupling with cyclopropylboronic acid at a mild 60°C. Crucially, this step achieves high conversion without the need for excessive reagents or toxic additives, and the product is isolated via simple recrystallization rather than chromatography. This shift dramatically simplifies the operation and reduces the generation of hazardous waste.
Subsequent steps involve nucleophilic aromatic substitution with nitro-anilines, reduction of the nitro group using Raney nickel, and final acryloylation to install the warhead group. Each transformation is tuned for maximum efficiency; for instance, the reduction step utilizes a THF/ethanol solvent system that facilitates easy filtration and catalyst recovery. The entire sequence is designed with commercial scale-up of complex pharmaceutical intermediates in mind, ensuring that every intermediate can be purified to >99% HPLC purity using crystallization. This approach not only secures the supply of critical high-purity EGFR inhibitor precursors but also aligns with modern green chemistry principles, offering a sustainable path forward for API manufacturing.
Mechanistic Insights into Copper-Catalyzed Cyclopropanation
The heart of this synthetic innovation lies in the optimized Chan-Lam-Evans type coupling used to install the cyclopropyl group on the indole nitrogen. Unlike traditional methods that require stoichiometric amounts of copper and harsh bases, this patent employs a catalytic system comprising copper acetate and 2,2'-bipyridine in the presence of sodium carbonate. The mechanism likely involves the transmetallation of the cyclopropyl group from boron to the copper center, followed by coordination with the indole nitrogen and subsequent reductive elimination. The use of bipyridine as a ligand stabilizes the copper species, allowing the reaction to proceed efficiently at 60°C in THF or acetonitrile. This lower temperature profile minimizes side reactions such as homocoupling of the boronic acid or decomposition of the sensitive pyrimidine ring.
Impurity control is rigorously managed through the choice of solvent and workup procedure. The patent specifies that the crude product can be directly recrystallized from ethanol, effectively removing residual copper species and unreacted starting materials without the need for silica gel. This is a critical advantage for reducing lead time for high-purity pharmaceutical intermediates, as chromatography is a major bottleneck in process development. Furthermore, the molar ratio of cyclopropylboronic acid is tightly controlled between 1:1 and 1:1.2, preventing the accumulation of boron-containing impurities that are difficult to remove. The result is a robust process capable of delivering intermediates with purity levels exceeding 99.6%, ensuring that downstream reactions proceed with high fidelity and yield.
How to Synthesize 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenyl Pyrimidine-2-amine Efficiently
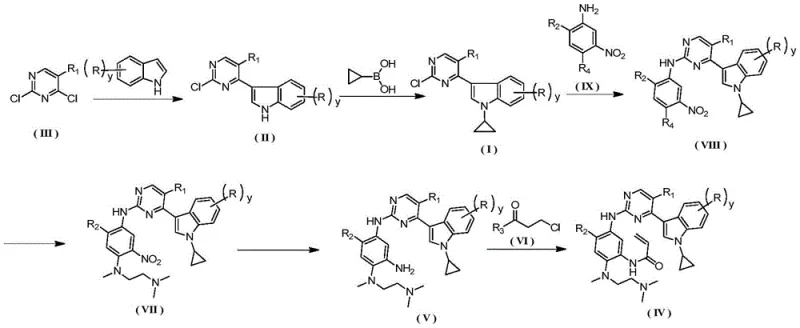
The synthesis of the target compound involves a convergent strategy that builds complexity step-by-step while maintaining strict control over reaction parameters. The process begins with the formation of the indole-pyrimidine core, followed by functionalization of the aniline moiety and final installation of the acrylamide group. Each step has been validated to ensure reproducibility and scalability, with specific attention paid to temperature control and reagent addition rates to manage exotherms. The detailed standardized synthesis steps below outline the precise conditions required to achieve the high yields and purities reported in the patent data.
- React 2,4-dichloropyrimidine with indole using a Grignard reagent to form the 3-(2-chloropyrimidin-4-yl)-1H-indole intermediate.
- Perform copper-catalyzed coupling with cyclopropylboronic acid at 60°C to introduce the cyclopropyl group without silica gel purification.
- Execute sequential nucleophilic substitutions, nitro reduction, and final acryloylation to obtain the target acryloylamide derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this new preparation method offers transformative advantages for procurement and supply chain operations. By shifting the starting point to commodity chemicals like indole and dichloropyrimidine, the dependency on niche, single-source suppliers for advanced intermediates is eliminated. This diversification of the raw material base significantly enhances supply chain resilience, mitigating the risk of production stoppages due to raw material shortages. Moreover, the simplification of the purification process—from column chromatography to recrystallization—drastically reduces the consumption of silica gel and organic solvents, leading to substantial cost savings in both materials and waste disposal.
- Cost Reduction in Manufacturing: The elimination of silica gel purification and the use of catalytic rather than stoichiometric amounts of expensive reagents directly lower the cost of goods sold (COGS). The ability to run reactions at lower temperatures (60°C vs 95°C) also reduces energy consumption. Furthermore, the high yield of each step minimizes the loss of valuable intermediates, ensuring that the overall process mass intensity (PMI) is optimized for economic efficiency without compromising quality.
- Enhanced Supply Chain Reliability: Sourcing readily available starting materials ensures a consistent and reliable supply flow. The robust nature of the chemistry means that batch-to-batch variability is minimized, which is critical for maintaining regulatory compliance. The simplified workup procedures allow for faster turnaround times between batches, enabling manufacturers to respond more agilely to market demand fluctuations and reducing the overall lead time for delivering high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process is inherently scalable, having been designed to avoid unit operations that do not translate well to large reactors, such as flash chromatography. The reduction in toxic solvents like toluene and the removal of highly toxic catalysts like DMAP align with increasingly stringent environmental regulations. This 'green' profile not only reduces the environmental footprint but also simplifies the permitting process for new manufacturing facilities, facilitating faster capacity expansion.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for decision-making. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this new method for their own production lines.
Q: How does this new synthesis route improve upon prior art methods for EGFR inhibitors?
A: The novel route eliminates the need for difficult-to-source raw materials like 3-(2-chloropyrimidin-4-yl)-1-cyclopropyl-1H-indole and replaces toxic solvents like toluene and catalysts like DMAP with safer, more efficient alternatives, significantly lowering production costs and environmental impact.
Q: What are the purity specifications achievable with this preparation method?
A: The process utilizes recrystallization instead of silica gel chromatography, consistently achieving HPLC purities exceeding 99% for key intermediates and the final active pharmaceutical ingredient, meeting stringent regulatory requirements.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the method features mild reaction conditions (e.g., 60°C coupling), simplified workup procedures, and reduced waste generation, making it highly scalable and compliant with modern green chemistry standards for commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-(1-cyclopropyl-1H-indol-3-yl)-N-phenyl Pyrimidine-2-amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the development of life-saving oncology drugs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab bench to manufacturing plant is seamless. We are committed to delivering stringent purity specifications for all our intermediates, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis or established catalog products, our infrastructure is designed to support your long-term supply needs with unwavering reliability.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis method can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits of switching to this more efficient route. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project requirements, ensuring that your development timeline remains on track.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →