Scalable Synthesis of EGFR Inhibitor Intermediates: A Catalyst-Free Route for Commercial Manufacturing
Scalable Synthesis of EGFR Inhibitor Intermediates: A Catalyst-Free Route for Commercial Manufacturing
The pharmaceutical landscape for oncology treatments is continuously evolving, with Epidermal Growth Factor Receptor (EGFR) inhibitors playing a pivotal role in treating Non-Small Cell Lung Cancer (NSCLC). A recent technological breakthrough detailed in patent CN111875539A introduces a highly efficient preparation method for (E)-N-[4-(3-ethynylphenyl)amino-3-cyano-7-ethoxy-6-quinolyl]-4-(dimethylamino)-2-butenamide, a potent irreversible pan-ErbB receptor tyrosine kinase inhibitor. This document outlines a sophisticated synthetic pathway that addresses critical bottlenecks in traditional manufacturing, offering a streamlined approach that enhances both chemical purity and process economics. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, understanding the nuances of this catalyst-free methodology is essential for securing a stable supply chain for next-generation antitumor therapies.
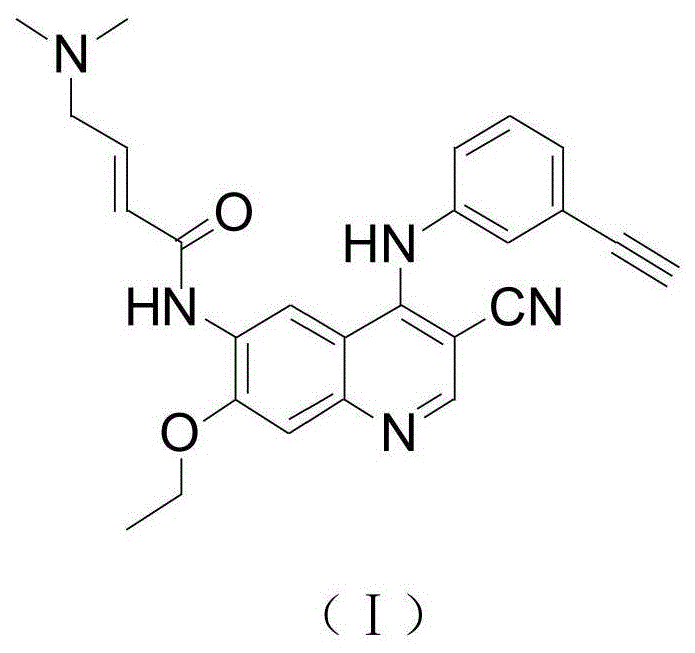
The core innovation lies in the strategic redesign of the synthetic route to eliminate dependency on harsh catalysts while maintaining exceptional yield metrics. By leveraging specific solvent systems and optimized reaction conditions, this method achieves a level of operational simplicity that translates directly into cost reduction in pharmaceutical intermediate manufacturing. The following analysis dissects the technical advantages of this route, providing a comprehensive view for stakeholders evaluating the feasibility of integrating this intermediate into their commercial production pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those reported in CN105175331A, typically rely on methanesulfonic acid as a catalyst to drive the substitution reaction between the quinoline core and the aniline derivative. While functional, these conventional approaches suffer from significant drawbacks that hinder industrial scalability and cost-efficiency. The presence of strong acid catalysts often leads to low conversion rates during the reaction process, necessitating extensive downstream purification to remove residual catalysts and associated by-products. Furthermore, the impurity content of the resulting intermediate, specifically Compound IV, is notoriously high, making quality control a arduous and expensive task. The difficulty in managing these impurity profiles not only extends the production cycle time but also increases the risk of batch failure, creating substantial volatility in the supply of high-purity pharmaceutical intermediates required for clinical and commercial applications.
The Novel Approach
In stark contrast, the novel methodology disclosed in the patent utilizes a catalyst-free substitution reaction mediated by n-propanol as the sole solvent. This shift represents a paradigm change in process chemistry, removing the need for expensive and corrosive acid catalysts entirely. The reaction between N-(4-chloro-3-cyano-7-ethoxyquinoline-6-yl)acetamide and m-aminophenylacetylene proceeds smoothly under heating conditions, typically between 90-98°C, achieving yields as high as 96% with purity levels exceeding 99.5%. This simplification of the reaction matrix not only accelerates the reaction kinetics but also dramatically simplifies the workup procedure, which often involves mere centrifugation and washing. The elimination of the catalyst removal step inherently reduces the environmental footprint and operational costs, positioning this route as a superior choice for cost reduction in electronic chemical manufacturing and broader fine chemical sectors.
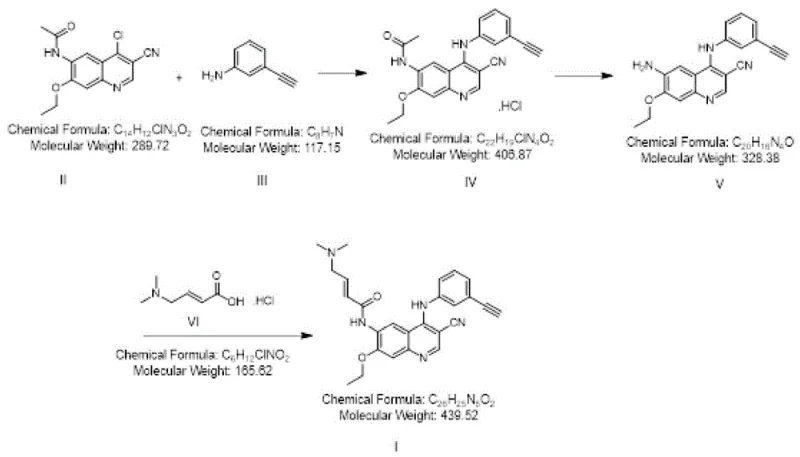
The robustness of this new approach is further evidenced by its successful validation across various reactor scales, from small laboratory flasks to 200L industrial kettles, demonstrating excellent reproducibility. The ability to consistently produce Compound IV with minimal impurities sets a strong foundation for the subsequent synthetic steps, ensuring that the final API intermediate meets the stringent specifications demanded by global regulatory bodies. This reliability is a critical factor for supply chain heads who prioritize continuity and predictability in their raw material sourcing strategies.
Mechanistic Insights into Catalyst-Free Nucleophilic Substitution
The mechanistic elegance of this synthesis centers on the nucleophilic aromatic substitution where the chlorine atom at the 4-position of the quinoline ring is displaced by the amino group of m-aminophenylacetylene. In traditional acid-catalyzed pathways, the protonation of the nitrogen atoms can sometimes lead to unwanted side reactions or decomposition of sensitive functional groups like the ethynyl moiety. By operating in a neutral n-propanol environment at elevated temperatures, the reaction leverages the intrinsic nucleophilicity of the aniline derivative without the interference of strong acids. This温和 condition preserves the integrity of the alkyne group, which is crucial for the biological activity of the final EGFR inhibitor. The choice of n-propanol is particularly critical; comparative experiments revealed that substituting it with ethanol or isopropanol resulted in inferior outcomes, characterized by higher impurity loads and reduced yields, highlighting the specific solvation effects that n-propanol provides to stabilize the transition state.
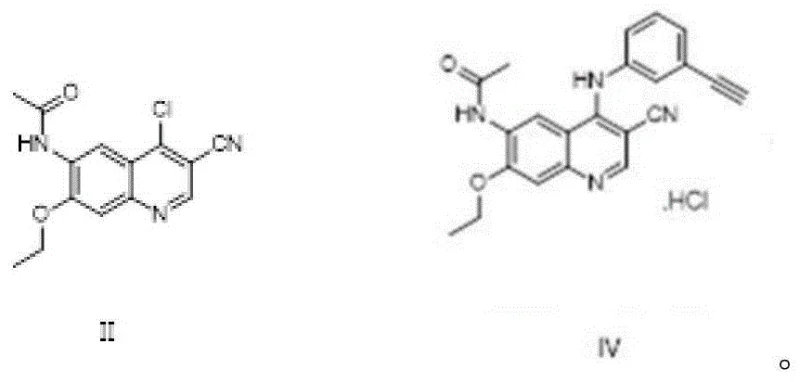
Following the formation of Compound IV, the subsequent hydrolysis to generate the free amine (Compound V) is executed with precise control over acid concentration and addition sequences. The patent specifies a two-stage reaction where concentrated hydrochloric acid is added first, followed by water, a sequence that proved vital for minimizing the formation of hydrolysis by-products. This controlled hydrolysis ensures that the acetamido protecting group is cleaved efficiently without degrading the sensitive quinoline-cyano framework. The final condensation step involves activating trans-4-dimethylamino crotonate with oxalyl chloride to form an acyl chloride intermediate, which then reacts with Compound V. Conducting this condensation at low temperatures (-20 to 0°C) in N-methyl pyrrolidone prevents polymerization and ensures the stereochemical integrity of the double bond, yielding the final (E)-isomer with high fidelity.
Impurity control is woven into every stage of this mechanism. By avoiding the methanesulfonic acid catalyst, the generation of sulfonate ester impurities is completely eradicated. Additionally, the specific temperature ramps and cooling protocols described, such as cooling the reaction mixture to 0-5°C before separation, facilitate the crystallization of the desired product while keeping soluble impurities in the mother liquor. This thermodynamic control over crystallization is a key driver for achieving the reported purity levels of over 99%, satisfying the rigorous demands of R&D directors focused on impurity谱 analysis and regulatory filing requirements.
How to Synthesize (E)-N-[4-(3-ethynylphenyl)amino-3-cyano-7-ethoxy-6-quinolyl]-4-(dimethylamino)-2-butenamide Efficiently
The synthesis of this complex EGFR inhibitor intermediate requires strict adherence to the optimized parameters regarding solvent ratios, temperature gradients, and reagent addition orders to maximize yield and purity. The process is divided into three distinct operational phases: the initial substitution to form the quinoline core, the hydrolytic deprotection to reveal the reactive amine, and the final amide coupling to install the side chain. Each phase has been engineered to minimize waste and energy consumption while ensuring safety and scalability. For process chemists looking to implement this route, the detailed standardized synthesis steps provided below offer a clear roadmap from raw materials to the final high-value intermediate.
- Perform a heating substitution reaction on N-(4-chloro-3-cyano-7-ethoxyquinoline-6-yl)acetamide and m-aminophenylacetylene in n-propanol solvent at 90-98°C to obtain Compound IV.
- Hydrolyze Compound IV under acidic conditions using concentrated hydrochloric acid and water in n-propanol, followed by neutralization with potassium carbonate to yield Compound V.
- Activate trans-4-dimethylamino crotonate with oxalyl chloride, then condense with Compound V in N-methyl pyrrolidone at low temperatures to finalize Compound I.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented synthesis route offers transformative advantages that directly address the pain points of procurement managers and supply chain directors. The primary benefit stems from the drastic simplification of the process workflow, which eliminates multiple unit operations associated with catalyst handling and removal. This streamlining translates into significant operational expenditure savings, as the facility requirements are less demanding and the throughput per batch is increased. Furthermore, the reliance on readily available commodity chemicals such as n-propanol, oxalyl chloride, and commercially sourced starting materials (Compounds II, III, and VI) mitigates the risk of raw material shortages. This accessibility ensures a stable and continuous supply chain, reducing the lead time for high-purity pharmaceutical intermediates and allowing manufacturers to respond more agilely to market demands.
- Cost Reduction in Manufacturing: The elimination of methanesulfonic acid and other transition metal catalysts removes the necessity for expensive scavenging resins or complex extraction protocols typically required to meet heavy metal limits. This qualitative shift in process design inherently lowers the cost of goods sold (COGS) by reducing reagent costs and waste disposal fees. Additionally, the high yield of 96% achieved in the key substitution step means that less raw material is wasted, maximizing the atom economy of the process. The simplified workup, often requiring only centrifugation and washing rather than column chromatography or extensive recrystallization, further reduces labor and utility costs, driving substantial cost savings in the overall manufacturing budget.
- Enhanced Supply Chain Reliability: The robustness of the n-propanol mediated reaction ensures consistent batch-to-batch quality, which is critical for maintaining long-term supply contracts with major pharmaceutical companies. The process has been demonstrated to scale effectively from laboratory benchtops to 200L reactors without loss of efficiency, indicating a low technical risk for commercial scale-up of complex pharmaceutical intermediates. By utilizing a solvent system that is easy to recover and recycle, the process also insulates the supply chain from volatile solvent markets. This stability allows procurement teams to forecast inventory needs with greater accuracy and secure reliable long-term pricing agreements with suppliers.
- Scalability and Environmental Compliance: The catalyst-free nature of the reaction significantly reduces the generation of hazardous waste streams, aligning the process with increasingly stringent global environmental regulations. The absence of heavy metals simplifies the effluent treatment process, lowering the environmental compliance burden on manufacturing sites. Moreover, the reaction conditions, primarily involving heating and stirring without the need for high-pressure equipment or cryogenic temperatures (except for the final step), make the process highly adaptable to existing multipurpose chemical plants. This ease of integration facilitates rapid capacity expansion, ensuring that supply can be scaled up quickly to meet surging demand for oncology therapeutics without requiring massive capital investment in new infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and comparative examples provided in the patent documentation, offering clarity on the practical benefits of adopting this technology. Understanding these details is crucial for technical teams evaluating the feasibility of this method for their specific production environments.
Q: Why is n-propanol preferred over ethanol or isopropanol in the substitution step?
A: Experimental data indicates that using ethanol or isopropanol results in significantly higher impurity profiles and lower yields compared to n-propanol. The specific solvation properties of n-propanol facilitate the nucleophilic substitution without requiring harsh acidic catalysts, leading to a product purity exceeding 99%.
Q: How does this method improve impurity control compared to prior art?
A: Traditional methods utilizing methanesulfonic acid catalysts often suffer from low conversion rates and difficult quality control due to complex impurity spectra. This novel approach eliminates the catalyst entirely and optimizes the hydrolysis sequence by controlling the addition order of hydrochloric acid and water, drastically reducing residual starting materials and side products.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process has been validated in reactor volumes up to 200L with consistent yields around 96%. The reliance on commercially available raw materials, simple workup procedures like centrifugation, and the absence of expensive transition metal catalysts make it highly robust for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable EGFR Inhibitor Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the development of life-saving oncology drugs. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to implement catalyst-free and environmentally friendly processes like the one described in CN111875539A demonstrates our dedication to innovation and sustainability in chemical manufacturing.
We invite procurement leaders and R&D directors to collaborate with us to optimize their supply chains for EGFR inhibitor production. By leveraging our technical expertise, we can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments, ensuring that your project moves forward with the highest level of confidence and efficiency. Let us be your partner in navigating the complexities of modern pharmaceutical synthesis.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →