Advanced Synthesis of Bioactive Benzotriazole Derivatives for Commercial Scale-up and Drug Development
The pharmaceutical industry is constantly seeking robust synthetic routes for novel heterocyclic compounds that exhibit potent biological activity, particularly in the realm of oncology. Patent CN101928254A introduces a significant advancement in this field by disclosing a series of benzotriazole derivatives characterized by a unique structural framework linking a benzotriazole core with various substituted benzoic acid moieties. These compounds, defined by the general formula where A, R, and R' can be hydrogen, hydroxyl, or methoxy groups, have demonstrated strong anti-proliferative activity against human oral epithelial cancer cells (KB), lung cancer cells (H460), and gastric cancer cells (MKN45). The innovation lies not only in the biological potential of these molecules but also in the efficiency of their preparation, which utilizes mild coupling conditions that are highly attractive for industrial application. By leveraging 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) as a coupling agent, the described methodology avoids the harsh conditions typically associated with traditional acylation reactions, thereby preserving sensitive functional groups and ensuring high product integrity.
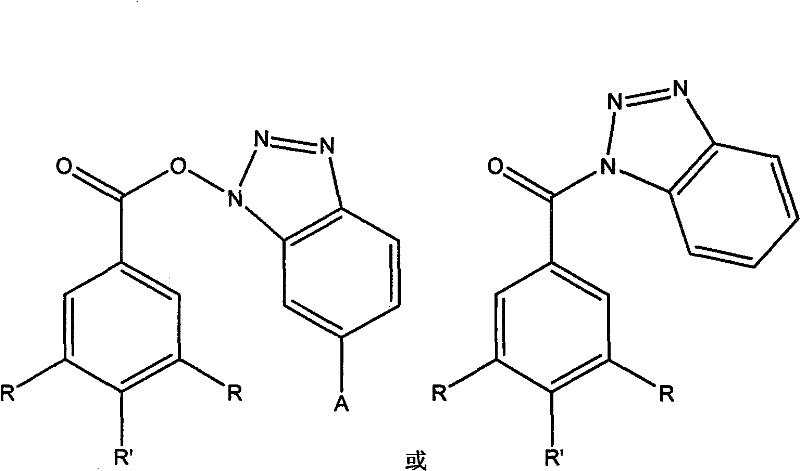
For research and development directors focusing on process chemistry, understanding the limitations of conventional synthesis methods is crucial for evaluating the value of this new technology. Traditionally, the formation of ester or amide bonds involving benzotriazole scaffolds often requires the conversion of carboxylic acids into highly reactive acid chlorides using reagents like thionyl chloride or oxalyl chloride. These classical approaches frequently necessitate elevated temperatures, strict anhydrous conditions, and generate corrosive gaseous byproducts that complicate waste management and equipment maintenance. Furthermore, the use of acid chlorides can lead to side reactions, particularly when the substrate contains other nucleophilic sites, resulting in complex impurity profiles that are difficult and costly to purge during downstream processing. The reliance on such aggressive reagents also poses safety hazards in large-scale manufacturing environments, increasing the operational risk and regulatory burden for pharmaceutical producers aiming to bring new anticancer agents to market.
In stark contrast, the novel approach detailed in the patent utilizes a carbodiimide-mediated coupling strategy that fundamentally shifts the paradigm towards greener and safer chemistry. By employing EDC·HCl in dichloromethane at room temperature, the reaction proceeds efficiently over a 24-hour period without the need for external heating or cooling systems. This mild condition is particularly advantageous for substrates containing thermally labile groups, such as the hydroxyl and methoxy substituents found in the preferred embodiments of this invention. The mechanism involves the in situ activation of the carboxylic acid to form an O-acylisourea intermediate, which is then attacked by the nucleophilic nitrogen or oxygen of the benzotriazole derivative. This pathway minimizes the formation of hazardous waste and simplifies the workup procedure, as the urea byproduct formed from EDC is generally water-soluble or easily removed via standard purification techniques like column chromatography. Consequently, this method offers a cleaner reaction profile and higher atom economy compared to traditional acid chloride routes.
Mechanistic Insights into EDC-Mediated Coupling for Benzotriazole Functionalization
To fully appreciate the technical depth of this synthesis, one must examine the mechanistic nuances of the EDC-catalyzed reaction. The process begins with the nucleophilic attack of the carboxylate oxygen on the central carbon of the carbodiimide group in EDC·HCl, generating a reactive O-acylisourea species. This activated intermediate is electrophilic enough to be attacked by the weakly nucleophilic nitrogen atom of the benzotriazole ring (in the case of amide formation) or the oxygen atom of the benzotriazol-1-ol (in the case of ester formation). The presence of the benzotriazole leaving group in the intermediate stages can further facilitate the acylation by forming an active ester, which is less prone to rearrangement than the initial O-acylisourea. This dual-activation capability ensures high conversion rates even with sterically hindered substrates, such as 3,4,5-trimethoxybenzoic acid. The reaction's success at room temperature indicates a favorable activation energy barrier, allowing the transformation to occur spontaneously once the reagents are mixed, which is a hallmark of a well-optimized catalytic system suitable for sensitive pharmaceutical intermediates.
Impurity control is another critical aspect where this mechanism excels, directly addressing the concerns of quality assurance teams. In traditional high-temperature acylations, thermal degradation of the starting materials or the product often leads to polymeric tars or decomposition products that co-elute with the desired compound. However, the ambient temperature operation of the EDC method significantly suppresses these thermal degradation pathways. Additionally, the specificity of the carbodiimide activation reduces the likelihood of non-selective acylation on other parts of the molecule, provided that the stoichiometry is carefully controlled. The patent specifies a molar ratio of 1:1:1.2 for the benzotriazole compound, benzoic acid, and EDC·HCl respectively, which ensures that the activating agent is present in slight excess to drive the reaction to completion without overwhelming the system with unreacted reagents. Post-reaction purification via column chromatography using an ethyl acetate and petroleum ether system effectively separates the target benzotriazole derivatives from the urea byproduct and any unreacted starting materials, yielding white crystalline solids with high purity as confirmed by NMR and elemental analysis data.
How to Synthesize Benzotriazole Esters and Amides Efficiently
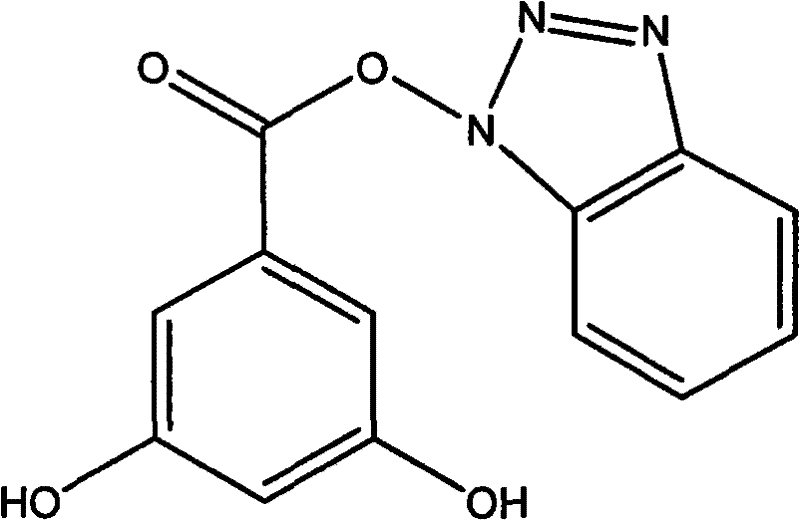
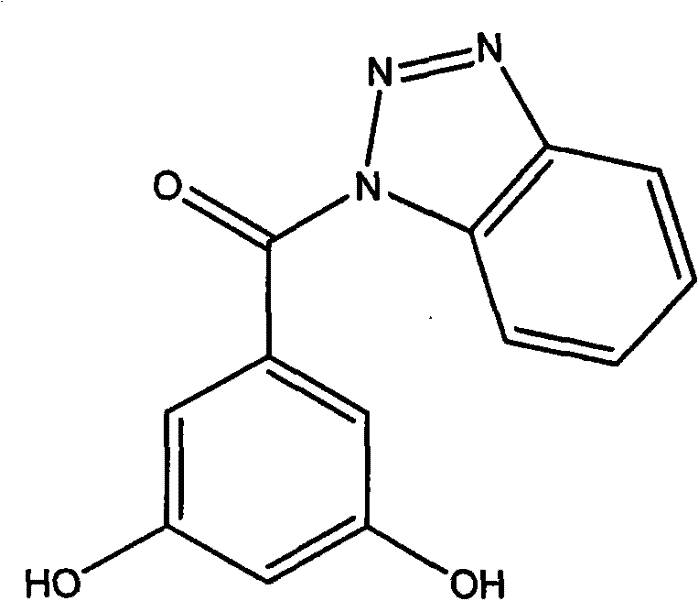
The practical implementation of this synthesis route is straightforward and relies on standard laboratory equipment, making it highly accessible for process development teams looking to scale up production. The procedure generally involves dissolving the benzotriazole precursor, such as 1H-benzo[d][1,2,3]-triazol-1-ol or its methoxy-substituted analogs, along with the chosen substituted benzoic acid in dichloromethane. Once the solution is homogeneous, the coupling agent EDC·HCl is added, and the mixture is stirred at room temperature for approximately 24 hours to ensure full conversion. Following the reaction, the solvent is removed under reduced pressure, and the crude residue is subjected to column chromatography to isolate the pure product. This standardized protocol has been successfully applied to a wide range of substrates, yielding compounds with melting points ranging from 96°C to 209°C and consistent spectral data, demonstrating the robustness and versatility of the method for generating diverse libraries of bioactive molecules.
- Preparation of the benzotriazole core: React o-nitrochlorobenzene with hydrazine hydrate in 1-heptanol at 110-120°C, followed by neutralization and recrystallization to obtain 1H-benzo[d][1,2,3]-triazol-1-ol.
- Activation and Coupling: Dissolve the benzotriazole intermediate and the substituted benzoic acid in dichloromethane, then add EDC·HCl catalyst at a molar ratio of 1:1:1.2.
- Purification: Stir the reaction mixture at room temperature for 24 hours, remove solvent under reduced pressure, and purify the crude product via column chromatography using ethyl acetate and petroleum ether.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain and procurement perspective, the adoption of this EDC-mediated synthesis route offers substantial strategic benefits that extend beyond mere chemical yield. The elimination of harsh reagents like thionyl chloride removes the need for specialized corrosion-resistant reactors and extensive scrubbing systems for acidic gases, leading to significant capital expenditure savings and reduced maintenance costs for manufacturing facilities. Furthermore, the ability to run reactions at room temperature drastically lowers energy consumption compared to processes requiring reflux or cryogenic cooling, contributing to a lower overall cost of goods sold (COGS). The use of common, commercially available solvents like dichloromethane and readily sourced starting materials such as o-nitrochlorobenzene and various benzoic acids ensures a stable and reliable supply chain, minimizing the risk of production delays due to raw material shortages. This stability is crucial for maintaining continuous production schedules in the fast-paced pharmaceutical sector.
- Cost Reduction in Manufacturing: The streamlined nature of this process eliminates multiple unit operations associated with traditional acid chloride synthesis, such as the separate preparation and distillation of acid chlorides. By combining the activation and coupling steps into a single pot at ambient temperature, manufacturers can reduce labor hours, utility usage, and reactor occupancy time. The high yields reported in the patent, often exceeding 75% and reaching up to 95% for intermediate steps, further enhance the economic viability by maximizing the output from each batch of raw materials. Additionally, the simplified purification process reduces the volume of silica gel and solvents required for chromatography, lowering waste disposal costs and environmental compliance burdens.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-stable reagents like EDC·HCl and solid benzoic acids mitigates the risks associated with handling volatile or moisture-sensitive liquids. This robustness allows for more flexible inventory management and reduces the likelihood of batch failures due to reagent degradation. The scalability of the method means that production can be easily ramped up from kilogram to tonne scales without significant process redesign, ensuring that supply can meet fluctuating market demands for anticancer drug intermediates. This flexibility is a key asset for contract development and manufacturing organizations (CDMOs) serving global pharmaceutical clients who require just-in-time delivery of high-quality intermediates.
- Scalability and Environmental Compliance: The green chemistry attributes of this method align perfectly with modern environmental regulations and corporate sustainability goals. By avoiding the generation of sulfur dioxide and hydrogen chloride gases, the process significantly reduces the facility's environmental footprint and simplifies permitting processes. The aqueous workup and standard organic solvent recovery systems are compatible with existing infrastructure in most chemical plants, facilitating easy technology transfer. Moreover, the high purity of the final products reduces the need for extensive recrystallization or reprocessing, further minimizing solvent waste and energy usage. This combination of operational efficiency and environmental stewardship makes the technology highly attractive for long-term commercial partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these benzotriazole derivatives, based on the detailed experimental data provided in the patent documentation. These insights are designed to clarify the feasibility of the process for industrial partners and highlight the specific advantages of the disclosed methodology over alternative synthetic routes. Understanding these details is essential for making informed decisions about integrating this technology into existing production pipelines for oncology drug development.
Q: What are the primary advantages of using EDC·HCl over traditional acid chloride methods for benzotriazole synthesis?
A: The use of EDC·HCl allows the reaction to proceed at room temperature without the need for harsh reagents like thionyl chloride, significantly reducing energy consumption and simplifying the removal of toxic byproducts, which is critical for pharmaceutical grade purity.
Q: How does the structural variation of the benzoic acid moiety affect the biological activity?
A: According to the patent data, substituents such as hydroxyl and methoxy groups on the benzoic acid ring significantly influence anti-proliferative activity against KB, H460, and MKN45 cancer cell lines, with specific patterns like 3,5-dihydroxy substitution showing potent IC50 values.
Q: Is this synthesis route scalable for industrial production of anticancer intermediates?
A: Yes, the process utilizes common solvents like dichloromethane and operates at ambient temperatures, avoiding extreme pressure or cryogenic conditions, which makes the commercial scale-up of complex benzotriazole derivatives highly feasible and safe.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzotriazole Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of next-generation anticancer therapeutics. Our team of expert chemists has extensively evaluated the technology described in CN101928254A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring these benzotriazole derivatives from the bench to the plant. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, utilizing our rigorous QC labs to ensure every batch conforms to the highest international standards. Our state-of-the-art facilities are equipped to handle the specific solvent systems and purification techniques outlined in the patent, guaranteeing a seamless transition from pilot scale to full commercial manufacturing.
We invite pharmaceutical companies and research institutions to collaborate with us to optimize their supply chains for these valuable bioactive compounds. By leveraging our expertise in process optimization and cost-effective manufacturing, we can help you achieve a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments for your projects. Let us be your partner in accelerating the development of life-saving medications through superior chemical engineering and reliable supply chain solutions.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →