Advanced Purification Technology for Pneumocandin B0 Enables Scalable Caspofungin Production
Advanced Purification Technology for Pneumocandin B0 Enables Scalable Caspofungin Production
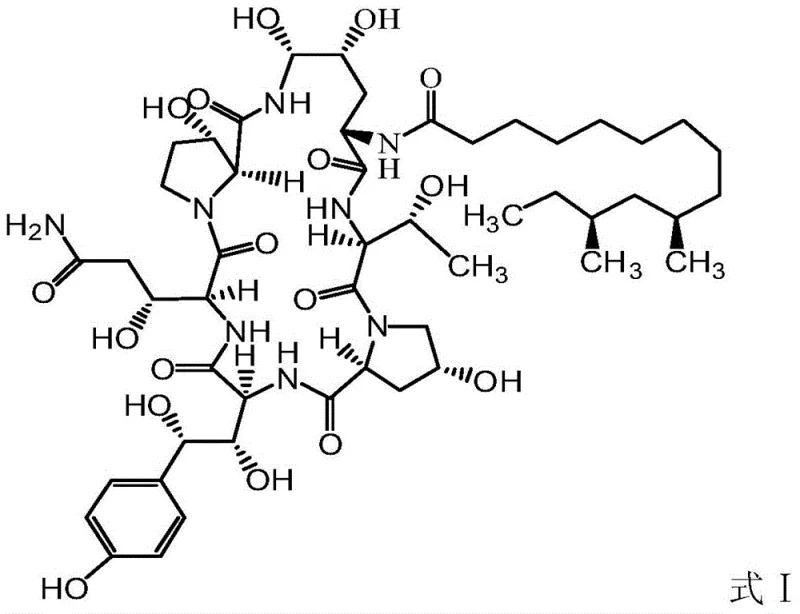
The pharmaceutical industry continuously seeks robust methodologies to enhance the purity and yield of critical active pharmaceutical ingredients, and the recent disclosures in patent CN105481952B offer a transformative approach to the production of Pneumocandin B0. This specific nitrogen-containing heterocyclic hexapeptide precursor serves as the fundamental building block for the synthesis of Caspofungin, a vital antifungal agent used in treating invasive infections. The patent details a sophisticated purification strategy that addresses the longstanding challenge of removing structural analog impurities, specifically the serine analog designated as Formula II, which traditionally compromises the quality of the final drug substance. By leveraging a novel suspension crystallization technique, this technology enables manufacturers to achieve unprecedented levels of purity without relying on costly and environmentally burdensome chromatographic methods. The implications for supply chain stability and cost efficiency are profound, as this method simplifies the downstream processing requirements significantly. For R&D directors and procurement specialists, understanding the mechanistic advantages of this process is essential for evaluating potential partnerships and optimizing production workflows. The ability to control impurity profiles at the intermediate stage directly correlates to the safety and efficacy of the final therapeutic product, making this innovation a cornerstone for modern antifungal manufacturing strategies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the purification of Pneumocandin B0 has been fraught with significant technical and economic hurdles, primarily due to the structural similarity between the target compound and its serine analog impurities. Conventional purification techniques, such as those described in prior art patents like WO2004042350A2 and WO0220618A1, heavily rely on preparative normal phase chromatography or macroporous adsorption resin methods. These traditional approaches are inherently inefficient because they often fail to reduce the content of the Formula II impurity to acceptable levels, frequently leaving residues greater than 2.5% in the final product. Furthermore, these chromatographic processes demand vast quantities of organic solvents, particularly acetonitrile, which poses severe environmental risks and escalates operational costs due to solvent recovery and disposal requirements. The reliance on high-pressure liquid chromatography also introduces bottlenecks in production throughput, making it difficult to scale operations to meet commercial demand without exponential increases in capital expenditure. Additionally, the use of toxic solvents necessitates stringent safety protocols and specialized equipment, further complicating the manufacturing landscape. For supply chain managers, these limitations translate into longer lead times, higher volatility in raw material costs, and increased regulatory scrutiny regarding environmental compliance and worker safety.
The Novel Approach
In stark contrast to the inefficiencies of chromatography, the method disclosed in CN105481952B introduces a paradigm shift by utilizing a controlled suspension crystallization process that exploits subtle solubility differences between the target molecule and its impurities. This innovative technique involves dissolving the crude Pneumocandin B0 in an aqueous organic solvent system where the water content is meticulously maintained between 8% and 30% by volume. Under these specific conditions, the target Formula I compound precipitates not as a traditional crystal, but as a coarse dispersion of non-crystalline solid particles with a diameter greater than 0.1 micrometers. Crucially, the serine analog impurity (Formula II) remains highly soluble in the solvent phase under these same conditions, allowing for a highly effective separation via simple filtration or centrifugation. This physical separation mechanism bypasses the need for complex resin interactions or high-pressure systems, drastically reducing the equipment footprint and energy consumption required for purification. The result is a streamlined process that achieves impurity levels below 0.1% with minimal loss of the target compound, typically less than 2%. This approach not only enhances product quality but also aligns with green chemistry principles by significantly reducing solvent usage and eliminating the need for toxic mobile phases often associated with preparative HPLC.
Mechanistic Insights into Suspension Crystallization Purification
The core scientific breakthrough of this technology lies in the precise manipulation of solubility parameters and particle physics within a multi-component solvent system. By adjusting the water content within the organic solvent matrix, specifically using alcohols like isobutanol or mixtures involving ethyl acetate, the process creates a thermodynamic environment where the saturation point of Pneumocandin B0 is exceeded while the impurity remains undersaturated. The formation of a coarse dispersion rather than a true crystalline lattice is critical, as it prevents the occlusion of impurities that often occurs during rapid crystal growth in traditional recrystallization. The patent specifies that the solid particles formed must have a diameter of at least 0.1 micrometers, a size range that facilitates efficient solid-liquid separation without clogging filtration media. This particle size distribution is monitored using laser diffraction techniques, ensuring consistency across batches. The mechanism effectively decouples the purification step from the chemical identity of the impurity, relying instead on physical state differences induced by solvent composition. This robustness makes the process less sensitive to minor variations in feedstock quality, providing a more reliable output for downstream synthesis. For technical teams, this means a more forgiving process window that maintains high purity standards even when starting material quality fluctuates within expected fermentation ranges.
Furthermore, the impurity control mechanism extends beyond simple physical separation to influence the quality of the final Caspofungin API. The serine analog impurity (Formula II) is known to carry through subsequent synthetic steps, converting into a structurally similar impurity in the final drug product that is difficult to remove later. By reducing the Formula II content to below 0.1% at the Pneumocandin B0 stage, the burden on the final purification steps is dramatically alleviated. This upstream control strategy ensures that the subsequent chemical modifications, such as the side-chain attachment and reduction steps required to form Caspofungin, proceed with higher fidelity. The reduction of impurity load early in the synthesis tree minimizes the formation of by-products that could complicate regulatory filings or require additional validation studies. From a quality assurance perspective, this mechanistic advantage provides a stronger control strategy for the overall manufacturing process, reducing the risk of batch failures due to out-of-specification impurity profiles. It represents a proactive approach to quality by design, where process parameters are tuned to prevent impurity formation or retention rather than relying solely on end-product testing.
How to Synthesize Pneumocandin B0 Efficiently
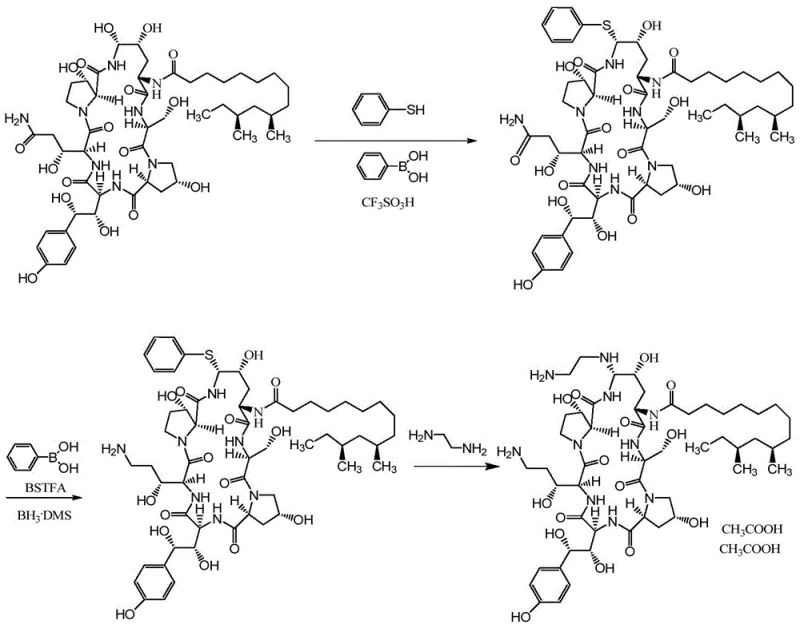
The implementation of this purification protocol requires careful attention to solvent selection and water content monitoring to ensure the formation of the desired suspension state. The process begins with dissolving the crude fermentation product in a suitable organic solvent system, followed by the controlled addition of an anti-solvent or temperature adjustment to induce precipitation. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility and compliance with the patent specifications. Adhering to these parameters is critical for achieving the target particle size and impurity reduction levels described in the technical data. Operators must ensure that the water content remains within the 8% to 30% window throughout the precipitation phase to maintain the coarse dispersion state. Deviations outside this range may result in crystallization or oiling out, which would compromise the purification efficiency. The following guide outlines the critical process parameters necessary for successful execution.
- Dissolve crude Pneumocandin B0 in an aqueous organic solvent solution with controlled water content.
- Induce precipitation by cooling or adding an anti-solvent to form a coarse dispersion suspension.
- Separate the suspended solid particles from the solvent via filtration or centrifugation to isolate high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this suspension crystallization technology offers substantial advantages in terms of cost structure and supply chain resilience. The elimination of preparative high-performance liquid chromatography (HPLC) removes a major cost driver associated with expensive resin columns and high-purity solvents like acetonitrile. Comparative data within the patent indicates that solvent consumption is drastically reduced, with the new method requiring significantly less volume per gram of product compared to traditional chromatographic purification. This reduction in solvent usage directly translates to lower raw material costs and reduced waste disposal fees, contributing to a more sustainable and economically viable manufacturing model. For procurement managers, this means a more stable cost base that is less susceptible to fluctuations in solvent markets. Additionally, the simplified equipment requirements allow for production in standard chemical processing facilities without the need for specialized high-pressure chromatography systems, lowering capital expenditure barriers for scale-up. The ability to produce high-purity intermediates consistently enhances supply reliability, ensuring that downstream API manufacturing schedules are not disrupted by purification bottlenecks or quality failures.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive preparative HPLC resins and reduces the volume of toxic organic solvents required for purification. By avoiding the use of large quantities of acetonitrile and specialized chromatography columns, the operational expenditure per kilogram of product is significantly lowered. The simplified workflow also reduces labor hours associated with column packing, maintenance, and solvent recovery, leading to overall efficiency gains. This cost structure allows for more competitive pricing of the final API while maintaining healthy margins for the manufacturer. The reduction in solvent waste also lowers environmental compliance costs, further enhancing the economic attractiveness of the technology.
- Enhanced Supply Chain Reliability: The robustness of the suspension crystallization method ensures consistent output quality even with variations in crude feedstock, reducing the risk of batch rejections. This reliability is crucial for maintaining continuous supply to downstream API manufacturers, preventing costly production stoppages. The scalability of the process means that production volumes can be increased to meet market demand without proportional increases in complexity or lead time. Suppliers utilizing this technology can offer more secure long-term contracts, knowing that their purification capacity is not limited by chromatographic throughput constraints. This stability is a key value proposition for pharmaceutical companies seeking to de-risk their supply chains for critical antifungal medications.
- Scalability and Environmental Compliance: The process is inherently scalable as it relies on standard unit operations like mixing, filtration, and drying rather than complex chromatographic separations. This makes it easier to transfer from pilot scale to commercial production without significant re-engineering. Environmentally, the reduction in hazardous solvent usage aligns with increasingly strict global regulations on volatile organic compound emissions and waste disposal. The ability to use less toxic solvent systems improves workplace safety and reduces the environmental footprint of the manufacturing site. These factors contribute to a stronger corporate sustainability profile, which is increasingly important for partnerships with major pharmaceutical companies committed to green chemistry initiatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this purification technology. These answers are derived directly from the experimental data and claims presented in the patent documentation to ensure accuracy and relevance. Understanding these details helps stakeholders evaluate the feasibility of integrating this method into their existing production workflows. The responses cover aspects of purity, scalability, and regulatory compliance to provide a comprehensive overview.
Q: How does this method remove serine analog impurities?
A: By controlling water content between 8% and 30%, Pneumocandin B0 forms a non-crystalline suspension while impurities remain dissolved in the solvent.
Q: What is the achievable purity level for the intermediate?
A: The process can reduce impurity content to below 0.1%, significantly improving the quality of the final Caspofungin product.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, it avoids expensive preparative HPLC and reduces solvent usage, making it highly viable for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pneumocandin B0 Supplier
NINGBO INNO PHARMCHEM stands at the forefront of implementing advanced purification technologies like the one described in CN105481952B to deliver high-quality pharmaceutical intermediates. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the benefits of this novel suspension crystallization method are fully realized at an industrial level. We maintain stringent purity specifications and operate rigorous QC labs to verify that every batch of Pneumocandin B0 meets the exacting standards required for Caspofungin synthesis. Our commitment to process innovation allows us to offer intermediates with superior impurity profiles, reducing the burden on our clients' downstream processing. By partnering with us, you gain access to a supply chain that is both cost-effective and technically robust, capable of supporting your long-term commercial goals.
We invite you to engage with our technical procurement team to discuss how this purification technology can optimize your specific manufacturing requirements. Request a Customized Cost-Saving Analysis to understand the potential economic impact of switching to our high-purity intermediates. Our experts are ready to provide specific COA data and route feasibility assessments tailored to your production needs. Let us help you streamline your supply chain and enhance the quality of your final antifungal products through our advanced manufacturing capabilities.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →