Advanced Synthesis of 2-Debenzoyl-2-Acyl Paclitaxel Derivatives for Commercial Oncology Applications
Advanced Synthesis of 2-Debenzoyl-2-Acyl Paclitaxel Derivatives for Commercial Oncology Applications
The pharmaceutical landscape for oncology treatments continues to evolve, driven by the relentless pursuit of compounds with superior efficacy and manageable toxicity profiles. Patent CN1108653A introduces a groundbreaking methodology for the preparation of 2-debenzoyl-2-acyl paclitaxel derivatives, representing a significant leap forward in the semi-synthesis of taxane-based anticancer agents. This technology addresses the longstanding chemical challenge of selectively modifying the C-2 position of the taxane nucleus, a region historically difficult to access without compromising the integrity of the complex molecular scaffold. By enabling the precise replacement of the native benzoyl group with diverse acyl moieties, this innovation opens new avenues for optimizing pharmacokinetic properties and enhancing antitumor potency. For global supply chain leaders and R&D directors, understanding this synthetic pathway is crucial for securing a reliable API intermediate supplier capable of delivering next-generation chemotherapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the chemical modification of paclitaxel has been fraught with difficulties due to the molecule's structural complexity and the presence of multiple reactive hydroxyl groups. Conventional attempts to alter the C-2 position often resulted in non-selective reactions, leading to mixtures of products that were arduous to separate and purify. The proximity of the C-2 benzoyl group to other sensitive functionalities, such as the C-1 acetate and the C-2' hydroxyl on the side chain, meant that harsh reaction conditions frequently caused degradation or unwanted epimerization. Furthermore, traditional methods lacked the finesse required to introduce bulky or electronically distinct acyl groups at the C-2 position without affecting the critical oxetane ring or the ester linkages essential for biological activity. These limitations severely restricted the ability of medicinal chemists to explore the structure-activity relationship (SAR) at this specific locus, thereby hindering the discovery of analogs with improved therapeutic indices.
The Novel Approach
The methodology disclosed in CN1108653A circumvents these historical bottlenecks through a sophisticated strategy of temporary protection and selective hydrolysis. By employing protecting groups such as tert-butyloxycarbonyl (t-BOC) or triethylsilyl (TES) at the C-2' and C-7 positions, the process effectively masks these reactive sites, rendering them inert during subsequent transformation steps. This strategic masking allows for the unprecedented selective cleavage of the C-2 benzoyl group using mild basic conditions, specifically lithium hydroxide, which leaves the rest of the molecule intact. Once the C-2 hydroxyl is exposed, it can be re-acylated with a wide variety of carboxylic acids or activated derivatives to install novel substituents. This approach not only ensures high regioselectivity but also maintains the stereochemical integrity of the taxane core, providing a robust platform for generating diverse libraries of potent analogs.
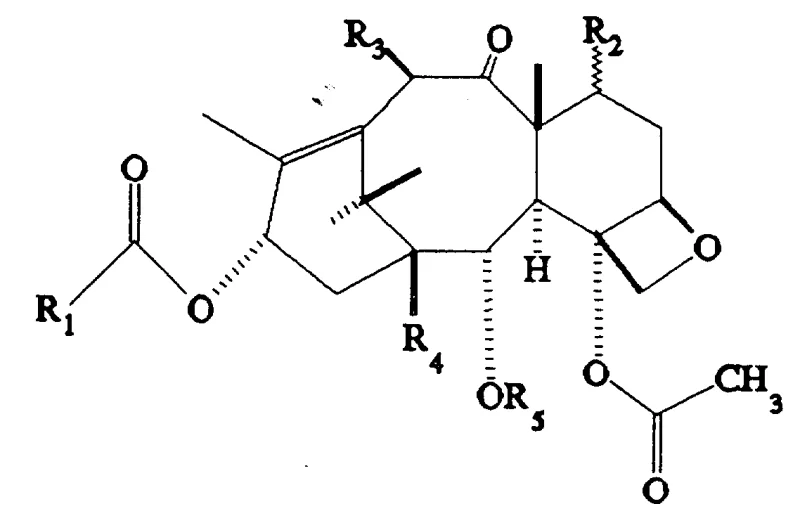
Mechanistic Insights into Selective Deacylation and Reacylation
The core of this technological advancement lies in the nuanced understanding of steric and electronic effects within the taxane framework. The mechanism begins with the protection of the C-2' amine and hydroxyl groups, often utilizing di-tert-butyl dicarbonate in the presence of a catalyst like DMAP. This step is critical as it prevents the nucleophilic attack on the side chain during the harsh hydrolysis phase. Following protection, the molecule undergoes a controlled hydrolysis where the C-2 benzoyl ester is selectively cleaved. The patent highlights the surprising finding that under specific conditions, the C-2 benzoyl group is more susceptible to hydrolysis than the C-4 acetate or the C-10 acetate when the C-7 and C-2' positions are shielded. This selectivity is the linchpin of the entire process, allowing for the generation of the 2-debenzoyl intermediate in high purity.
Subsequent reacylation involves reacting the newly freed C-2 hydroxyl with activated acylating agents, such as acid chlorides or anhydrides, often facilitated by coupling reagents like DCC. The versatility of this step allows for the introduction of aryl, alkyl, or heteroaryl groups, enabling fine-tuning of the molecule's lipophilicity and binding affinity to tubulin. Finally, the removal of the protecting groups is achieved under mild acidic conditions, typically using formic acid, which restores the native functionality of the side chain and the C-7 hydroxyl without inducing elimination or rearrangement reactions. This meticulous orchestration of protection, reaction, and deprotection ensures that the final product retains the essential pharmacophore while benefiting from the enhanced properties conferred by the C-2 modification.
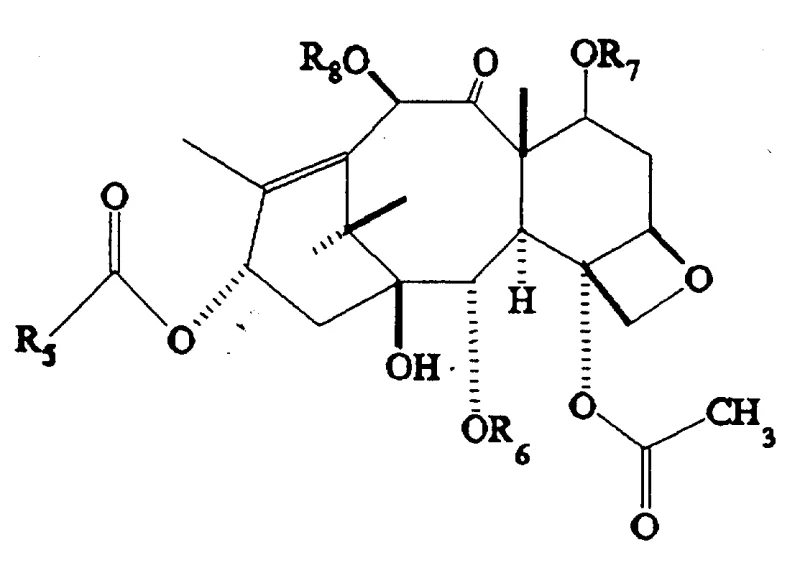
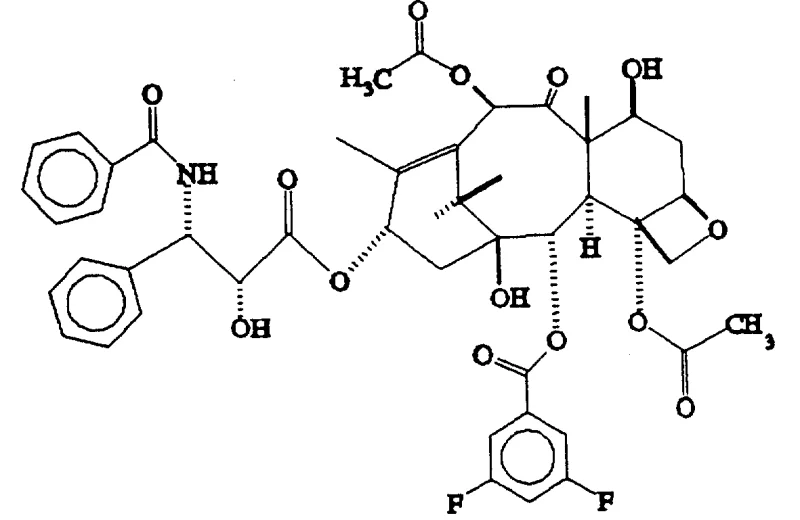
Certain embodiments of this chemistry have yielded compounds with extraordinary biological profiles. For instance, the introduction of electron-withdrawing groups on the C-2 benzoyl ring has been shown to dramatically increase cytotoxicity. The patent specifically cites the 3,5-difluorobenzoyl derivative as exhibiting activity approximately 25,000 times greater than paclitaxel in specific in vivo assays against P-388 leukemia. This staggering increase in potency underscores the importance of the C-2 position in modulating drug-target interactions and suggests that even minor structural changes can lead to major therapeutic breakthroughs. Such findings validate the utility of this synthetic route for developing high-purity paclitaxel analogs that could potentially overcome multidrug resistance mechanisms observed in clinical settings.
How to Synthesize 2-Debenzoyl-2-Acyl Paclitaxel Efficiently
The practical implementation of this synthesis requires precise control over reaction parameters to maximize yield and minimize impurities. The process generally initiates with the dissolution of paclitaxel in an anhydrous solvent such as acetonitrile or DMF, followed by the addition of the protecting group reagent. Careful monitoring via TLC or HPLC is essential to ensure complete protection before proceeding to the hydrolysis step. The hydrolysis itself must be conducted at controlled temperatures, often near 0°C initially, to prevent over-hydrolysis of other ester linkages. Following the isolation of the 2-debenzoyl intermediate, the acylation step utilizes standard peptide coupling conditions or acid chloride chemistry, depending on the stability of the desired acyl group. The final deprotection is a delicate balance of acidity and time to ensure clean removal of silyl or carbamate groups without degrading the sensitive taxane core.
- Protect the C-2' and C-7 hydroxyl groups of paclitaxel using tert-butyloxycarbonyl (t-BOC) or triethylsilyl (TES) groups to prevent unwanted side reactions.
- Perform selective hydrolysis of the C-2 benzoyl group using lithium hydroxide (LiOH) under controlled conditions to generate the 2-debenzoyl intermediate.
- React the 2-debenzoyl intermediate with specific acylating agents (such as substituted benzoic acids) to introduce the desired acyl group at the C-2 position.
- Remove the protecting groups using formic acid or mild acidic conditions to yield the final 2-debenzoyl-2-acyl paclitaxel derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling strategic benefits beyond mere chemical elegance. The ability to derivatize paclitaxel at the C-2 position using semi-synthetic methods significantly reduces reliance on complex total synthesis, which is often cost-prohibitive and low-yielding. By starting from readily available paclitaxel or baccatin III precursors, manufacturers can leverage existing supply chains for the core taxane nucleus while adding value through downstream chemical modification. This modular approach facilitates cost reduction in pharmaceutical manufacturing by streamlining the production of high-value analogs that command premium pricing in the oncology market. Furthermore, the use of common reagents like lithium hydroxide, DCC, and formic acid ensures that the process is easily scalable using standard industrial equipment, mitigating the need for specialized or exotic catalysts that could introduce supply bottlenecks.
- Cost Reduction in Manufacturing: The elimination of complex total synthesis steps and the utilization of semi-synthetic pathways from abundant natural precursors drastically lower the raw material costs associated with producing advanced taxane analogs. By focusing on late-stage functionalization, the process minimizes the number of purification steps required for the core scaffold, thereby reducing solvent consumption and waste generation. This efficiency translates directly into a more favorable cost of goods sold (COGS), allowing for competitive pricing strategies in the global API market without compromising on quality or purity standards.
- Enhanced Supply Chain Reliability: The robustness of the protection-deprotection strategy ensures consistent batch-to-batch reproducibility, a critical factor for maintaining uninterrupted supply to downstream drug product manufacturers. The reliance on stable, commercially available reagents reduces the risk of supply disruptions caused by the scarcity of specialized chemicals. Additionally, the flexibility of the acylation step allows for the rapid adaptation of the synthesis to produce different analogs based on market demand, providing supply chain agility that is essential in the fast-paced pharmaceutical industry.
- Scalability and Environmental Compliance: The reaction conditions described are amenable to large-scale production, with hydrolysis and acylation steps that can be safely managed in standard reactor vessels. The process avoids the use of heavy metal catalysts or highly toxic reagents, aligning with modern green chemistry principles and simplifying regulatory compliance regarding residual impurities. This environmental compatibility not only reduces the burden of waste treatment but also enhances the sustainability profile of the manufacturing operation, a key consideration for increasingly eco-conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these novel paclitaxel derivatives. Understanding these details is vital for partners evaluating the feasibility of integrating these intermediates into their development pipelines. The answers are derived directly from the technical specifications and experimental data provided in the underlying patent documentation, ensuring accuracy and relevance for decision-makers.
Q: How does the biological activity of 2-debenzoyl-2-acyl paclitaxel compare to standard paclitaxel?
A: According to patent CN1108653A, specific derivatives such as the 3,5-difluorobenzoyl analog exhibit significantly enhanced antitumor activity, reported to be up to 25,000 times more active than paclitaxel in certain in vivo models against P-388 leukemia.
Q: What is the key chemical challenge addressed by this synthesis method?
A: The primary challenge is the regioselective modification of the C-2 position on the taxane B-ring without affecting other sensitive functional groups like the C-2' hydroxyl or the oxetane D-ring. This method solves it through specific protecting group strategies.
Q: Are these derivatives suitable for improving water solubility?
A: Yes, one of the stated objectives of the invention is to produce paclitaxel analogs with improved water solubility compared to taxol, facilitating better bioavailability and formulation options for cancer treatment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Debenzoyl-2-Acyl Paclitaxel Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the technologies described in CN1108653A for the future of oncology therapeutics. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory discovery to market availability is seamless. Our state-of-the-art facilities are equipped to handle the stringent purity specifications required for oncology APIs, supported by rigorous QC labs that employ advanced analytical techniques to verify identity and potency. We are committed to delivering high-purity paclitaxel analogs that meet the exacting standards of global regulatory bodies, providing you with a secure and reliable source for your critical drug substances.
We invite you to engage with our technical procurement team to discuss how our capabilities align with your specific project requirements. Whether you need a Customized Cost-Saving Analysis for your current supply chain or require specific COA data and route feasibility assessments for new analogs, our experts are ready to assist. By partnering with us, you gain access to a wealth of chemical expertise and manufacturing capacity designed to accelerate your development timelines and reduce your overall operational risks. Contact us today to explore how we can support your mission to bring life-saving cancer treatments to patients worldwide.