Advanced Manufacturing Of High-Purity Orlistat Via Catalytic Hydrogenation And Chromatography
Advanced Manufacturing Of High-Purity Orlistat Via Catalytic Hydrogenation And Chromatography
The pharmaceutical industry constantly seeks robust, scalable, and cost-effective pathways for producing high-value active pharmaceutical ingredients (APIs) and their intermediates. A significant breakthrough in this domain is documented in Chinese patent CN102304105A, which details a novel method for preparing high-purity Orlistat. This technology moves away from the capital-intensive constraints of traditional preparative high-performance liquid chromatography (HPLC) and instead leverages a sophisticated combination of mixed silica gel medium-pressure chromatography and precise crystallization techniques. For R&D directors and supply chain leaders, this represents a pivotal shift towards more sustainable and economically viable manufacturing. The process begins with Lipstatin fermentation broth and navigates through a series of optimized unit operations including leaching, extraction, decolorization, and a critical catalytic hydrogenation step. By achieving a total yield greater than 30% and a final product purity exceeding 99.5% with single impurities below 0.1%, this methodology sets a new benchmark for quality and efficiency in the production of anti-obesity agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the purification of Orlistat to meet stringent International Council for Harmonisation (ICH) standards has been fraught with technical and economic challenges. Traditional total synthesis routes, such as those disclosed in earlier patents like CN 1319596A, often involve cumbersome reaction sequences extending up to twelve steps, resulting in low overall yields and exorbitant reagent costs. Furthermore, while semi-synthetic approaches starting from Lipstatin fermentation exist, the purification stage has typically relied on dynamic axial compression (DAC) preparative columns or reverse-phase liquid chromatography (RPLC). Although these methods can achieve high purity, they suffer from severe limitations regarding industrial scalability. The equipment investment for DAC and RPLC is prohibitively high, the packing materials are expensive, and the batch processing capacity is inherently small, making them unsuitable for the multi-ton production volumes required by the global market. Additionally, these conventional methods often struggle to consistently maintain single impurity levels below the critical 0.1% threshold without incurring massive operational expenses.
The Novel Approach
The innovative strategy outlined in patent CN102304105A effectively dismantles these barriers by introducing a streamlined process centered on mixed silica gel medium-pressure chromatography. This approach replaces the need for ultra-high-pressure systems with robust, standard chemical equipment that is widely available and significantly cheaper to install and maintain. The core of this novelty lies in the specific packing of the chromatography column, utilizing a gradient of silica gel mesh sizes (e.g., combining 100-200 mesh and 200-300 mesh) to optimize separation efficiency without the high backpressure associated with fine particles. Coupled with a carefully controlled elution system using ethyl acetate and acetone, this method allows for the processing of large volumes of Lipstatin concentrate. Following chromatography, the process employs a multi-stage crystallization and crystal form conversion protocol. This ensures not only chemical purity but also the physical stability of the final polymorph. The result is a manufacturing route capable of producing 1.5 to 3 tons per month, demonstrating a clear path to commercial scale-up that was previously unattainable with older purification technologies.
Mechanistic Insights into Pd/C Catalyzed Hydrogenation
At the heart of the chemical transformation from Lipstatin to Orlistat lies a critical catalytic hydrogenation step, which is elegantly depicted in the reaction scheme below. This step involves the saturation of specific double bonds within the Lipstatin structure to yield the tetrahydro-derivative, Orlistat. The patent specifies the use of palladium on carbon (Pd/C) as the heterogeneous catalyst, a choice driven by its high activity and selectivity under mild conditions. The reaction is conducted in a hydrogenation reactor where hydrogen gas is introduced at pressures ranging from 0.1 MPa to 1.0 MPa, with an optimal preference around 0.5 MPa. Temperature control is equally vital, maintained between 20°C and 50°C, preferably at 40°C, to prevent side reactions or degradation of the sensitive beta-lactone ring. The mechanistic efficiency here is paramount; the catalyst facilitates the addition of hydrogen across the olefinic bonds without affecting the ester or lactone functionalities, ensuring the structural integrity of the pharmacophore is preserved. This selectivity is crucial for maintaining the biological activity of the final drug substance.
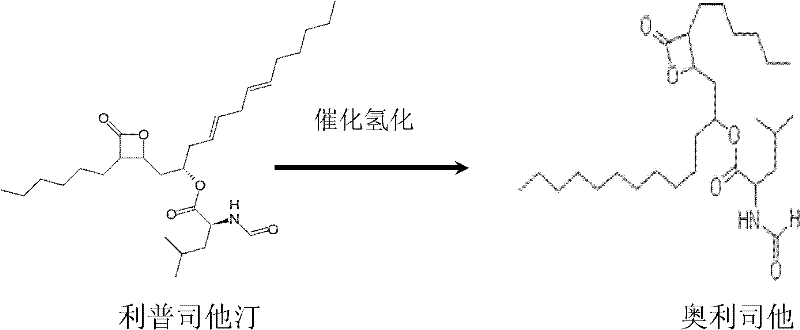
Beyond the primary transformation, the mechanistic design of this process includes rigorous impurity control measures integrated directly into the workflow. Prior to hydrogenation, the Lipstatin intermediate undergoes a decolorization step using activated carbon, which removes colored impurities and potential catalyst poisons that could inhibit the hydrogenation reaction. Post-hydrogenation, the removal of the Pd/C catalyst via filtration ensures no heavy metal residues remain in the product, a critical compliance factor for pharmaceutical safety. The subsequent crystallization steps act as a final polishing mechanism. By manipulating solvent polarity, temperature, and anti-solvent addition rates, the process selectively precipitates the desired Orlistat crystal lattice while leaving residual impurities in the mother liquor. The final crystal form conversion step, utilizing solvents like heptane or ether, guarantees the formation of the thermodynamically stable polymorph required for consistent dissolution profiles and bioavailability in the final dosage form. This holistic approach to mechanism and purification ensures that the final product consistently meets the >99.5% purity specification.
How to Synthesize Orlistat Efficiently
Implementing this synthesis route requires precise adherence to the optimized parameters established in the patent data to ensure reproducibility and high yield. The process begins with the leaching of fermented mycelium, where the ratio of biomass to solvent is critical for maximizing Lipstatin recovery. Following extraction and concentration, the medium-pressure chromatography step must be carefully monitored for flow rates and eluent composition to achieve the necessary separation of Lipstatin from fermentation byproducts. The hydrogenation reaction demands strict safety protocols regarding hydrogen handling, while the crystallization phases require precise thermal control to induce nucleation and growth of the correct crystal habit. For a detailed, step-by-step breakdown of the operational parameters, including specific solvent ratios, temperature gradients, and filtration protocols, please refer to the standardized synthesis guide provided below.
- Leach Lipstatin from fermented mycelium using organic solvents like ethanol or acetone, followed by extraction with heptane or ethyl acetate to concentrate the active ingredient.
- Purify the concentrated Lipstatin using mixed silica gel medium-pressure chromatography with an ethyl acetate and acetone mobile phase to achieve purity greater than 93%.
- Perform catalytic hydrogenation using Pd/C catalyst at controlled pressure and temperature to convert Lipstatin to Orlistat, followed by multi-step crystallization and crystal form conversion.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this manufacturing technology offers profound strategic advantages that extend far beyond simple technical metrics. The shift from high-cost preparative HPLC to medium-pressure chromatography using common chemical equipment drastically reduces the capital expenditure (CAPEX) required for facility setup. This lower barrier to entry translates directly into a more competitive cost structure for the final API, allowing for better margin management in a price-sensitive market. Furthermore, the use of recyclable solvents such as ethanol, ethyl acetate, and heptane aligns with modern green chemistry principles, significantly reducing waste disposal costs and environmental compliance burdens. The ability to recover and reuse these solvents creates a closed-loop system that minimizes raw material consumption and mitigates the risk of supply chain disruptions caused by volatile solvent markets.
- Cost Reduction in Manufacturing: The elimination of expensive chromatography fillers and high-pressure pumping systems results in substantial operational cost savings. By utilizing standard stainless steel reactors and columns, maintenance costs are minimized, and the lifespan of the equipment is extended. The process avoids the use of toxic or hazardous reagents found in total synthesis routes, further reducing the costs associated with specialized handling and waste treatment. This economic efficiency makes the production of high-purity Orlistat financially sustainable even at large volumes, providing a reliable cost advantage over competitors relying on older, less efficient purification technologies.
- Enhanced Supply Chain Reliability: The scalability of this process is a key driver for supply chain stability. With a demonstrated production capacity of 1.5 to 3 tons per month, manufacturers can confidently meet large-volume orders without the bottlenecks typical of batch-limited HPLC processes. The reliance on fermentation-derived Lipstatin ensures a renewable starting material source, decoupling production from the fluctuations of petrochemical feedstocks often used in total synthesis. Additionally, the robustness of the crystallization steps ensures high recovery rates, minimizing material loss and maximizing the output from each batch of fermentation broth. This reliability is essential for maintaining continuous supply to downstream formulation partners.
- Scalability and Environmental Compliance: The technology is inherently designed for industrial amplification, moving seamlessly from pilot scale to multi-ton commercial production. The use of common chemical equipment means that scaling up does not require bespoke engineering solutions, reducing the time-to-market for new capacity. From an environmental perspective, the process supports clean production goals through solvent recycling and the avoidance of heavy metal contaminants in the final product. This alignment with environmental, social, and governance (ESG) criteria enhances the brand value of the supply chain and ensures long-term regulatory compliance in strict markets like the US and Europe.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Orlistat using this patented methodology. These insights are derived directly from the experimental data and beneficial effects reported in the patent literature, providing a transparent view of the process capabilities. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this supply source into their existing manufacturing networks. The answers reflect the balance between high-purity standards and practical industrial application.
Q: What is the purity level achievable with this Orlistat synthesis method?
A: The patented process utilizing mixed silica gel chromatography and crystallization achieves an HPLC purity of over 99.5%, with single impurities controlled below 0.1%, meeting strict ICH standards for pharmaceutical applications.
Q: How does this method improve upon traditional preparative HPLC purification?
A: Unlike traditional preparative HPLC which requires expensive equipment and has limited batch processing capabilities, this method uses common chemical equipment and medium-pressure chromatography, allowing for industrial scale-up of several tons per month with significantly lower investment costs.
Q: Is the catalytic hydrogenation step safe for large-scale production?
A: Yes, the hydrogenation step operates at moderate pressures between 0.1 MPa and 1.0 MPa and temperatures between 20°C and 50°C using standard hydrogenation reactors, ensuring safety and feasibility for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Orlistat Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced manufacturing technologies to secure the global supply of essential pharmaceutical intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patents like CN102304105A are fully realized in practice. We operate stringent purity specifications and maintain rigorous QC labs to verify that every batch of Orlistat meets the >99.5% purity and <0.1% single impurity thresholds required by international pharmacopoeias. Our commitment to technical excellence ensures that our clients receive a product that is not only chemically superior but also consistent in its physical properties, facilitating smooth downstream processing.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By leveraging our optimized catalytic hydrogenation and chromatography platforms, we can help you reduce lead time for high-purity pharmaceutical intermediates while securing a stable, long-term supply. Contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our engineering expertise can drive value for your organization.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →