Scalable Synthesis of Gamma-Carboline Derivatives via Rhodium-Catalyzed C-H Activation for Commercial Applications
Scalable Synthesis of Gamma-Carboline Derivatives via Rhodium-Catalyzed C-H Activation for Commercial Applications
The pharmaceutical and fine chemical industries are constantly seeking efficient pathways to access complex heterocyclic scaffolds, particularly those with significant biological potential. Patent CN112979648A introduces a groundbreaking methodology for the synthesis of gamma-carboline derivatives, a class of compounds increasingly recognized as vital bioisosteres to beta-carbolines. Unlike traditional methods that often suffer from harsh conditions and limited substrate scope, this invention leverages a sophisticated rhodium-catalyzed C-H activation strategy to construct the tricyclic core from simple indole precursors. The process is characterized by its "de novo" construction approach, utilizing widely available raw materials to achieve high reaction efficiency and structural diversity. For R&D directors and procurement specialists, this represents a significant opportunity to streamline the supply chain for neuroactive and antitumor candidate molecules, ensuring a reliable source of high-purity intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of gamma-carboline derivatives has been plagued by significant technical and economic bottlenecks that hinder large-scale commercialization. Conventional routes often rely on specialized starting materials containing pre-installed functional groups, such as amides or oxime ethers, which require multi-step preparation from basic commodities. These legacy methods frequently employ metal-catalyzed intramolecular cyclizations that demand rigorous exclusion of moisture and oxygen, alongside expensive ligands that drive up the cost of goods sold (COGS). Furthermore, many existing protocols result in N-substituted carboline products, which necessitate additional deprotection steps to unlock the nitrogen atom for downstream medicinal chemistry modifications. The cumulative effect of these inefficiencies is a prolonged development timeline, lower overall yields, and a supply chain vulnerable to raw material shortages, making it difficult to secure a reliable gamma-carboline supplier for clinical trial materials.
The Novel Approach
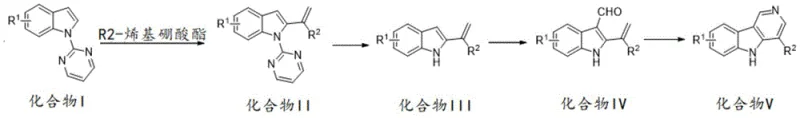
The methodology disclosed in CN112979648A fundamentally reshapes the manufacturing landscape by employing a direct C-H alkenylation strategy that bypasses the need for pre-functionalized substrates. By utilizing a pyrimidine group as a transient directing group on the indole nitrogen, the process achieves exceptional regioselectivity during the initial carbon-carbon bond formation. This innovative route allows for the use of simple indoles and alkenyl boronates, which are commodity chemicals with stable global supply chains. The subsequent steps involve mild deprotection, formylation, and a tandem cyclization that efficiently closes the third ring without requiring extreme temperatures or pressures. This streamlined four-step sequence not only simplifies the operational complexity but also significantly reduces waste generation, aligning with modern green chemistry principles and offering substantial cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Rhodium-Catalyzed C-H Alkenylation and Cyclization
The core of this synthetic breakthrough lies in the precise orchestration of transition metal catalysis, specifically utilizing a cationic rhodium(III) species generated in situ from [RhCp*Cl2]2 and silver acetate (AgOAc). In the initial C-H alkenylation step, the pyrimidine moiety coordinated to the indole nitrogen acts as a powerful directing group, guiding the rhodium catalyst to the C2 position of the indole ring. This coordination facilitates the activation of the inert C-H bond, allowing for the insertion of the alkenyl boronate ester with high fidelity. The presence of AgOAc serves a dual purpose: it acts as an oxidant to regenerate the active Rh(III) species and assists in the transmetallation process with the boronate. This mechanistic pathway ensures that the alkenyl group is installed exclusively at the desired position, minimizing the formation of regioisomers that would complicate downstream purification and compromise the purity specifications required for API intermediates.
Following the alkenylation, the removal of the pyrimidine directing group under basic conditions (NaOEt/DMSO) restores the indole NH functionality, which is crucial for the final cyclization event. The subsequent Vilsmeier-Haack formylation introduces an aldehyde moiety at the C3 position, setting the stage for the ring-closing reaction. The final transformation involves a tandem imidization and cyclization mediated by hydroxylamine hydrochloride. Mechanistically, the hydroxylamine condenses with the aldehyde to form an oxime intermediate, which then undergoes an intramolecular nucleophilic attack by the indole nitrogen onto the activated alkene side chain. This cascade reaction efficiently constructs the pyridine ring of the gamma-carboline scaffold in a single pot, demonstrating high atom economy. The robustness of this mechanism allows for a wide tolerance of substituents (R1 and R2), enabling the rapid generation of diverse libraries for structure-activity relationship (SAR) studies without the need for process re-optimization.
How to Synthesize Gamma-Carboline Derivatives Efficiently
The practical implementation of this synthesis route is designed for seamless translation from laboratory discovery to pilot plant operations. The process begins with the N-substitution of indole with 2-chloropyrimidine, followed by the critical rhodium-catalyzed coupling. Operators must maintain strict control over the molar ratios, particularly keeping the catalyst loading between 0.01 and 0.3 equivalents to balance cost and conversion rates. The reaction temperature for the alkenylation is optimized at 60°C in methanol, providing a safe and scalable thermal profile. Subsequent steps utilize common industrial solvents like DMSO and dioxane, ensuring compatibility with standard stainless steel reactors. The detailed standardized synthesis steps, including specific workup procedures and purification parameters, are outlined below to ensure reproducibility and quality consistency across batches.
- Perform C-H alkenylation of Compound I with R2-alkenyl boronate using [RhCp*Cl2]2 and AgOAc catalysts in MeOH at 60°C to obtain Compound II.
- Remove the pyrimidine directing group from Compound II using sodium ethoxide in DMSO at 100°C to yield Compound III.
- Conduct Vilsmeier-Haack formylation on Compound III using POCl3 and DMF, followed by NaOH workup to generate Compound IV.
- Execute tandem imidization and cyclization of Compound IV with hydroxylamine hydrochloride and sodium acetate in dioxane at 100°C to form the final gamma-carboline.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers compelling strategic advantages that extend beyond mere technical feasibility. The reliance on commodity starting materials like indole and styryl boronic acid esters mitigates the risk of supply disruptions often associated with exotic or custom-synthesized reagents. By eliminating the need for precious metal scavengers typically required in palladium-catalyzed cross-couplings, the process simplifies the purification workflow and reduces the burden on waste management systems. The high efficiency of the tandem cyclization step means fewer unit operations are required, directly translating to reduced utility consumption and shorter manufacturing cycles. These factors collectively contribute to a more resilient supply chain capable of meeting the demanding timelines of drug development programs while maintaining competitive pricing structures.
- Cost Reduction in Manufacturing: The elimination of expensive pre-functionalized starting materials and the use of low-loading rhodium catalysis significantly lower the raw material costs. Furthermore, the high selectivity of the C-H activation step reduces the formation of difficult-to-remove impurities, thereby decreasing the cost associated with chromatographic purification and increasing the overall yield of the final product. This economic efficiency makes the commercial scale-up of complex gamma-carboline derivatives financially viable for both generic and innovator pharmaceutical companies.
- Enhanced Supply Chain Reliability: By anchoring the synthesis on widely available indole derivatives, the method insulates the production schedule from the volatility of niche chemical markets. The robustness of the reaction conditions, which do not require cryogenic temperatures or ultra-high pressures, ensures that manufacturing can proceed reliably in diverse geographic locations. This flexibility allows for the establishment of redundant supply sources, reducing lead time for high-purity pharmaceutical intermediates and ensuring continuity of supply for critical clinical trials.
- Scalability and Environmental Compliance: The process utilizes solvents and reagents that are well-understood in terms of environmental health and safety (EHS) profiles, facilitating easier regulatory approval for manufacturing sites. The stepwise nature of the synthesis allows for intermediate isolation and quality control checks, ensuring that any deviations are caught early before valuable resources are committed to subsequent steps. This controlled approach supports sustainable manufacturing practices by minimizing solvent waste and energy consumption per kilogram of product produced.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of gamma-carboline derivatives using this patented methodology. These insights are derived directly from the experimental data and process descriptions within the patent documentation, providing clarity on the feasibility and advantages of this route for potential partners and stakeholders.
Q: What are the advantages of this gamma-carboline synthesis method over traditional routes?
A: This method utilizes commercially available indole as a starting material, avoiding complex multi-step precursor synthesis. It features mild reaction conditions, high regioselectivity via Rh-catalysis, and leaves the nitrogen atom unsubstituted for further derivatization.
Q: Is the rhodium catalyst system scalable for industrial production?
A: Yes, the process uses standard solvents like methanol and DMSO and operates at moderate temperatures (60-100°C). The catalyst loading is low (0.01-0.3 molar ratio relative to substrate), making it economically viable for large-scale manufacturing.
Q: How does this route impact the purity profile of the final API intermediate?
A: The stepwise approach with distinct purification stages (column chromatography or crystallization) between intermediates ensures high purity. The specific C-H activation mechanism minimizes side reactions, resulting in a cleaner impurity profile suitable for pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Gamma-Carboline Derivative Supplier
At NINGBO INNO PHARMCHEM, we understand that the transition from a patented laboratory method to a commercial reality requires deep technical expertise and robust infrastructure. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves smoothly from gram-scale optimization to full-scale manufacturing. Our facilities are equipped with state-of-the-art rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of gamma-carboline intermediate meets the highest standards required for pharmaceutical applications. We are committed to delivering quality and consistency, leveraging our technical prowess to navigate the complexities of organometallic catalysis and heterocyclic synthesis.
We invite you to collaborate with us to leverage this advanced synthesis technology for your drug development pipeline. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can accelerate your timeline to market while optimizing your overall project economics.