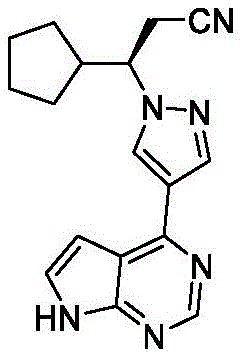
Advanced Synthesis of Ruxolitinib Intermediates: Scalable Solutions for Global API Manufacturing
The pharmaceutical industry continuously seeks robust pathways for the production of critical kinase inhibitors, and the synthesis of Ruxolitinib remains a focal point for generic drug manufacturers aiming to enter the myelofibrosis treatment market. Patent CN115850115A introduces a groundbreaking preparation method for a key Ruxolitinib intermediate, specifically targeting the efficient production of compounds with formula VI and V, ultimately leading to the high-purity chiral intermediate (S)-3-cyclopentyl-3-hydroxypropionitrile. This technical breakthrough addresses the longstanding challenges associated with low enantiomeric excess and costly separation techniques found in earlier literature. By leveraging a novel chiral resolution strategy rather than relying solely on asymmetric reduction, this method ensures an ee value of greater than 98 percent, setting a new benchmark for quality in pharmaceutical intermediate manufacturing. For global supply chain leaders, understanding this proprietary route is essential for securing a reliable source of high-quality raw materials that meet stringent regulatory standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Ruxolitinib precursors has been plagued by significant technical bottlenecks that hinder cost-effective mass production. Early approaches, such as those described in patent WO2010083283, relied heavily on Suzuki coupling reactions followed by conjugate addition, necessitating chiral column chromatography for separation. This dependency on chromatographic purification is economically prohibitive for industrial applications due to the high cost of chiral columns and the low throughput associated with batch processing. Furthermore, alternative resolution methods using agents like D-(+)-dibenzoyltartaric acid have demonstrated suboptimal results, often requiring multiple recrystallization cycles to achieve acceptable purity, which drastically reduces overall yield. Other reported methods involving R-CBS catalyzed reduction have failed to consistently deliver the required stereochemical purity, with experimental replicates showing ee values as low as 60 percent, far below the 98 percent threshold needed for API synthesis. These inefficiencies create substantial risks for procurement managers concerned with supply continuity and cost stability.
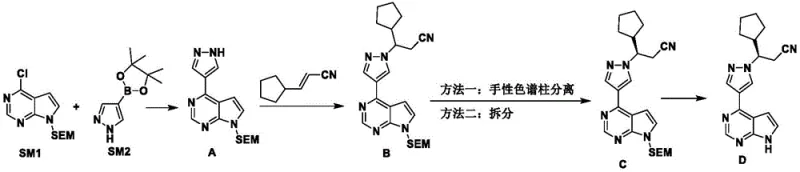
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in CN115850115A utilizes a sophisticated diastereomeric resolution strategy that bypasses the need for expensive chiral catalysts or preparative HPLC. The core innovation lies in the condensation of a racemic hydroxynitrile mixture with a specific resolving agent, N-acetyl-L-phenylalanine, to form separable diastereomers. This chemical transformation allows for the physical separation of the desired (S)-enantiomer through simple recrystallization in solvents such as methyl tert-butyl ether or dichloromethane. The process is designed for high material utilization, as the unwanted isomer can be recycled back into the synthesis loop through oxidation and reduction steps, minimizing waste generation. This approach not only simplifies the operational workflow but also significantly enhances the economic viability of producing high-purity pharmaceutical intermediates. By shifting from catalytic asymmetry to classical resolution with recycling, the method offers a more predictable and scalable solution for commercial scale-up of complex API intermediates.
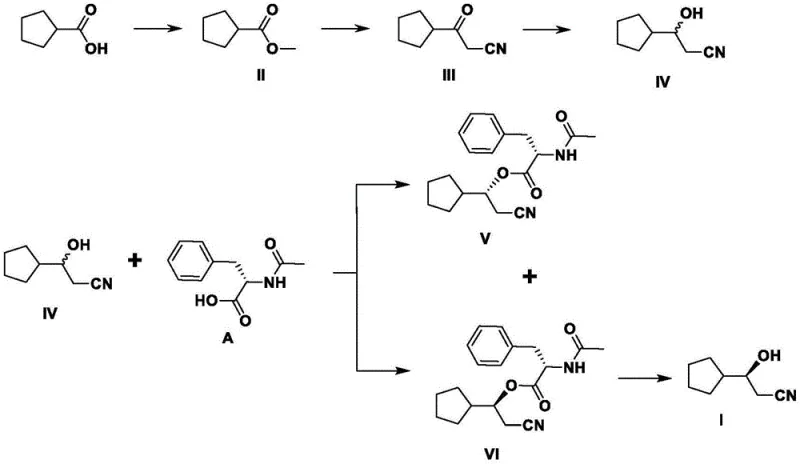
Mechanistic Insights into Chiral Resolution and Condensation
The heart of this synthetic advancement is the precise stereochemical control achieved during the condensation reaction between the racemic compound IV and the chiral resolving agent A. Mechanistically, the hydroxyl group of the racemic 3-cyclopentyl-3-hydroxypropionitrile reacts with the carboxylic acid moiety of the resolving agent in the presence of a condensing reagent like EDCI and a catalyst such as DMAP. This reaction generates two distinct diastereomeric esters, labeled as compound V and compound VI, which possess different physical properties, particularly solubility. The structural difference arises from the spatial arrangement of the cyclopentyl and cyanoethyl groups relative to the chiral center of the resolving agent. This disparity in physicochemical behavior is exploited during the recrystallization phase, where the less soluble diastereomer (Compound VI) precipitates out of the solution with high optical purity. The ability to tune this separation by adjusting solvent polarity and temperature is critical for R&D directors focused on impurity profiling and process robustness.
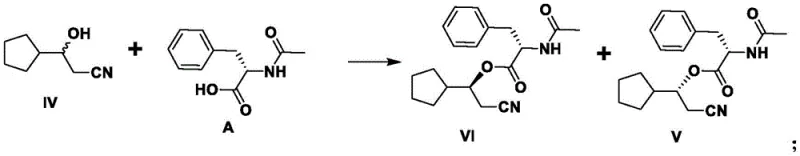
Furthermore, the process incorporates a closed-loop system for impurity management and material recovery, which is vital for maintaining consistent quality across batches. The mother liquor containing the undesired diastereomer (Compound V) is not discarded but instead subjected to hydrolysis to regenerate the racemic alcohol, which can then be oxidized back to the ketone precursor (Compound III) and re-entered into the reduction stream. This recycling capability ensures that the overall atom economy of the process is maximized, reducing the environmental footprint and raw material costs. Additionally, the resolving agent itself can be recovered from the aqueous hydrolysis phase by acidification, achieving recovery rates of approximately 90 percent with high purity. This level of process integration demonstrates a deep understanding of green chemistry principles, offering a sustainable pathway for cost reduction in pharmaceutical intermediate manufacturing while adhering to strict environmental compliance standards.
How to Synthesize Ruxolitinib Intermediate Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for laboratory and pilot-scale production, emphasizing precise control over reaction parameters to ensure reproducibility. The process begins with the preparation of the ketonitrile precursor via esterification and substitution, followed by a controlled reduction to generate the racemic alcohol. The critical resolution step requires careful management of stoichiometry, with a preferred molar ratio of resolving agent to substrate between 0.9 and 1.5, typically optimized at 1.2 equivalents. Reaction temperatures are maintained between -10°C and 25°C during the addition phase to prevent racemization, followed by a warming period to complete the condensation. Detailed standardized synthesis steps see the guide below.
- Perform esterification of cyclopentanecarboxylic acid followed by substitution with acetonitrile to form the ketonitrile precursor.
- Reduce the ketonitrile to a racemic hydroxynitrile mixture using sodium borohydride under controlled temperatures.
- Execute chiral resolution using N-acetyl-L-phenylalanine to separate diastereomers, followed by hydrolysis to obtain the target chiral intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers transformative benefits for organizations managing the procurement of complex organic intermediates. By eliminating the reliance on scarce and expensive chiral catalysts like R-CBS, the process inherently lowers the bill of materials and reduces exposure to supply chain volatility associated with specialized reagents. The ability to recover and reuse both the starting materials and the resolving agent creates a circular economy within the manufacturing process, leading to substantial cost savings over the lifecycle of the product. For supply chain heads, this translates to a more resilient sourcing strategy where raw material availability is less dependent on external niche suppliers. The simplified purification steps, which avoid complex chromatography, also reduce processing time and energy consumption, further enhancing the economic efficiency of the operation.
- Cost Reduction in Manufacturing: The elimination of chiral column chromatography and expensive transition metal catalysts removes significant cost centers from the production budget. The recycling of the resolving agent and the ability to convert the unwanted isomer back into the main synthesis stream drastically reduce raw material waste. This qualitative improvement in material efficiency directly correlates to a lower cost of goods sold, allowing for more competitive pricing strategies in the generic drug market without compromising on quality margins.
- Enhanced Supply Chain Reliability: The use of commodity chemicals such as cyclopentanecarboxylic acid, methanol, and common solvents like dichloromethane ensures that the supply chain is not vulnerable to shortages of exotic reagents. The robustness of the resolution chemistry means that production schedules are less likely to be disrupted by batch failures due to low enantiomeric excess. This reliability is crucial for maintaining continuous API production lines and meeting the demanding delivery timelines of downstream pharmaceutical customers.
- Scalability and Environmental Compliance: The process is designed with industrial scalability in mind, utilizing standard unit operations like crystallization, filtration, and liquid-liquid extraction that are easily transferred from pilot plants to multi-ton reactors. The reduced generation of hazardous waste, thanks to the recycling loops and avoidance of heavy metals, simplifies waste treatment protocols and ensures compliance with increasingly stringent environmental regulations. This makes the technology an attractive option for manufacturers looking to expand capacity while maintaining a strong sustainability profile.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. They are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the feasibility and advantages of the method. Understanding these details is key for technical teams evaluating the adoption of this new process for their manufacturing portfolios.
Q: What is the chiral purity achieved by this new resolution method?
A: The patented method efficiently prepares the target intermediate with an ee value exceeding 98%, significantly higher than the 60-63% typically observed in prior art reduction methods.
Q: Can the resolving agent be recovered to reduce costs?
A: Yes, the process includes a recovery step where the resolving agent is acidified from the aqueous phase, achieving a recovery yield of approximately 90% with high purity.
Q: Is this synthesis route suitable for industrial scale-up?
A: Absolutely. The method avoids expensive chiral column chromatography and uses common solvents like dichloromethane and methanol, making it highly viable for large-scale commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ruxolitinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic routes to maintain competitiveness in the global pharmaceutical market. Our team of expert chemists has thoroughly analyzed the technology disclosed in CN115850115A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are committed to delivering stringent purity specifications and operate rigorous QC labs to ensure that every batch of Ruxolitinib intermediate meets the highest international standards. Our infrastructure is uniquely positioned to translate this innovative laboratory method into a reliable, large-scale supply solution for your organization.
We invite you to collaborate with us to optimize your supply chain and reduce manufacturing costs through the implementation of this superior synthesis technology. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities align with your strategic goals for API production.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →