Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharma
The pharmaceutical landscape is continuously evolving, driven by the demand for more efficient synthetic routes to bioactive heterocycles. A significant breakthrough in this domain is detailed in patent CN113045503B, which discloses a robust preparation method for 2-trifluoromethyl substituted quinazolinone compounds. These scaffolds are pivotal in medicinal chemistry, serving as the core structure for numerous therapeutic agents ranging from antifungal and antiviral medications to anticancer drugs. The introduction of a trifluoromethyl group into these heterocyclic systems is particularly strategic, as it significantly enhances metabolic stability, lipophilicity, and bioavailability compared to their non-fluorinated counterparts. This patent presents a transition metal palladium-catalyzed carbonylation cascade reaction that utilizes cheap and readily available trifluoroethylimidoyl chloride and amines as starting materials, marking a substantial departure from legacy synthetic approaches.
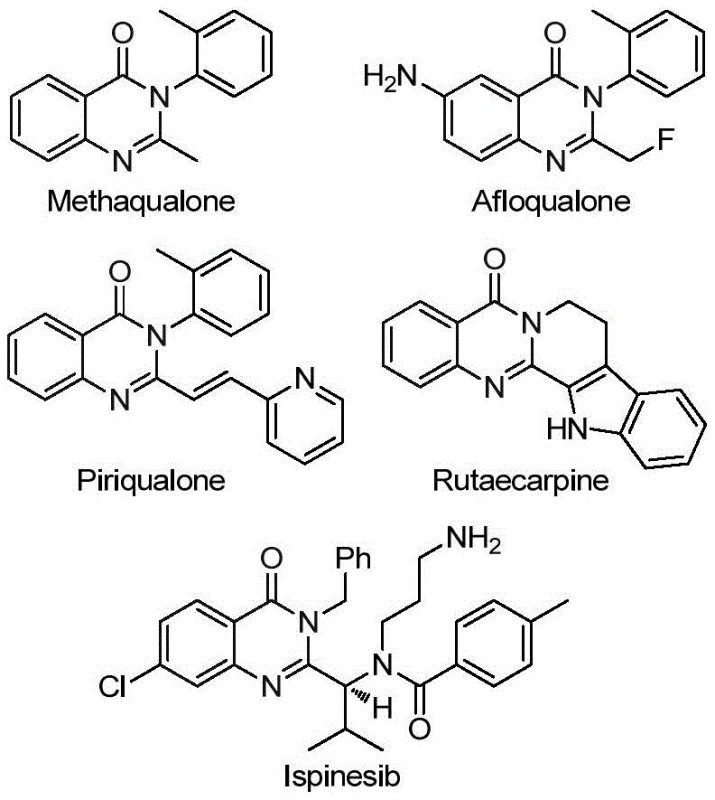
The versatility of this synthetic methodology is underscored by its applicability to a wide array of biologically active molecules. As illustrated in the structural diversity of known drugs, the quinazolinone core is a privileged scaffold found in compounds such as Methaqualone, Afloqualone, and the potent kinesin inhibitor Ispinesib. Furthermore, the natural product Rutaecarpine, known for its anti-inflammatory and vasorelaxant properties, also shares this fundamental architecture. The ability to efficiently construct these complex frameworks with high regioselectivity and functional group tolerance is a critical capability for any advanced chemical manufacturer aiming to support global drug development pipelines. The method described in CN113045503B not only simplifies the construction of these cores but also introduces the valuable trifluoromethyl motif in a single operational step, thereby streamlining the path from bench-scale discovery to commercial production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl-substituted quinazolinones has been fraught with significant technical and economic challenges that hinder large-scale manufacturing. Conventional literature reports typically rely on cyclization reactions involving anthranilamides with ethyl trifluoroacetate, trifluoroacetic anhydride, or trifluoroacetic acid under varying conditions. Alternatively, methods utilizing anthranilates with unstable trifluoroacetamides or isatoic anhydrides with trifluoroacetic anhydride have been employed. Another common approach involves T3P-promoted cascade reactions of anthranilic acid, trifluoroacetic acid, and amines. These traditional pathways are generally limited by harsh reaction conditions that require stringent temperature and pressure controls, often leading to safety concerns in industrial settings. Moreover, the substrates required, such as trifluoroacetic anhydride or specialized coupling reagents, are frequently expensive and may require pre-activation steps that add complexity and cost to the process. Perhaps most critically, these older methods often suffer from low yields and narrow substrate scopes, failing to accommodate diverse functional groups which limits their utility in the rapid synthesis of analog libraries for drug discovery.
The Novel Approach
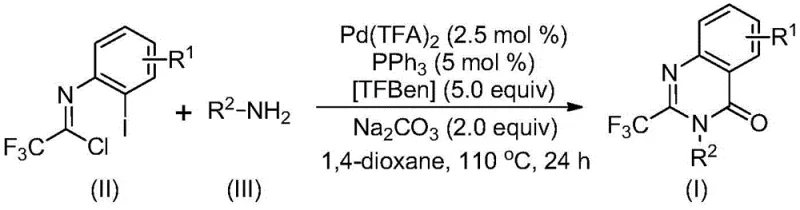
In stark contrast to these legacy methods, the novel approach disclosed in the patent utilizes a palladium-catalyzed carbonylation cascade that fundamentally reshapes the synthetic logic. By employing trifluoroethylimidoyl chloride and various amines as the primary building blocks, the process bypasses the need for expensive pre-activated carboxylic acid derivatives. The reaction proceeds efficiently in the presence of a palladium catalyst, a phosphine ligand, and a carbon monoxide substitute, specifically TFBen (1,3,5-tricarboxylic acid phenol ester). This strategy allows for the direct assembly of the quinazolinone ring system with the simultaneous incorporation of the trifluoromethyl group. The operational simplicity is a major advantage; the reaction can be conducted in common organic solvents like 1,4-dioxane at moderate temperatures. This new route demonstrates exceptional substrate compatibility, tolerating a wide range of substituents on both the aromatic ring and the amine component, including alkyl, halogen, and alkoxy groups. Consequently, this method provides a versatile platform for generating diverse libraries of 2-trifluoromethyl quinazolinones with high reaction efficiency and practical utility for industrial applications.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The success of this transformation relies on a sophisticated catalytic cycle orchestrated by the palladium center. The reaction likely initiates with a base-promoted intermolecular carbon-nitrogen bond coupling between the trifluoroethylimidoyl chloride and the amine, generating a trifluoroacetamidine derivative in situ. Subsequently, the palladium catalyst, generated from precursors like palladium trifluoroacetate and triphenylphosphine, undergoes oxidative addition into the carbon-iodine bond of the aromatic substrate. This forms a key divalent palladium intermediate. Crucially, the carbon monoxide required for the carbonylation is not supplied as a hazardous gas but is released in situ from TFBen under heating conditions. This generated CO then inserts into the carbon-palladium bond to form an acyl palladium intermediate. The presence of a base facilitates the formation of a palladium-nitrogen bond, leading to the generation of a seven-membered ring palladium intermediate. The cycle concludes with a reductive elimination step that releases the final 2-trifluoromethyl-substituted quinazolinone product and regenerates the active palladium catalyst. This mechanistic pathway ensures high atom economy and minimizes the formation of toxic byproducts associated with traditional carbonylation reagents.
Beyond the primary catalytic cycle, the method exhibits remarkable control over impurity profiles, which is paramount for pharmaceutical grade intermediates. The use of TFBen as a solid CO surrogate eliminates the risks associated with handling gaseous carbon monoxide, thereby reducing the potential for side reactions caused by uncontrolled CO pressure. Furthermore, the specific choice of ligands and the mild basic conditions provided by sodium carbonate help to suppress competitive hydrolysis of the imidoyl chloride or the final product. The reaction conditions are optimized to favor the intramolecular cyclization over intermolecular oligomerization, ensuring that the desired quinazolinone core is formed selectively. Post-reaction processing is equally streamlined; the crude mixture can be simply filtered to remove inorganic salts and palladium residues, followed by standard silica gel chromatography. This ease of purification suggests that the reaction generates minimal tarry byproducts or complex impurity mixtures, facilitating the isolation of high-purity material suitable for downstream biological testing or further chemical modification.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The practical implementation of this synthesis is designed for reproducibility and scalability, making it an ideal candidate for technology transfer from R&D to pilot plant operations. The protocol outlines a straightforward procedure where all reagents, including the palladium catalyst, ligand, CO surrogate, base, and substrates, are combined in a single vessel. The reaction is heated to 110°C for a duration of 16 to 30 hours, allowing sufficient time for the cascade sequence to reach completion. Detailed standardized synthesis steps for executing this protocol are provided in the guide below, ensuring that operators can achieve consistent results regardless of batch size. The robustness of the method is evidenced by its successful application in the synthesis of various derivatives, consistently delivering high yields across different substrate classes.
- Combine palladium trifluoroacetate, triphenylphosphine, TFBen, sodium carbonate, trifluoroethylimidoyl chloride, and amine in an organic solvent like 1,4-dioxane.
- Heat the reaction mixture at 110°C for 16 to 30 hours to facilitate the carbonylation cascade cyclization.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers tangible strategic benefits that extend beyond mere chemical elegance. The shift from expensive, pre-activated reagents to commodity chemicals like trifluoroethylimidoyl chloride and simple amines represents a fundamental optimization of the bill of materials. This transition directly addresses the perennial challenge of cost reduction in pharmaceutical intermediate manufacturing by lowering the raw material input costs significantly. Furthermore, the elimination of hazardous gaseous reagents simplifies the engineering controls required for production, potentially reducing capital expenditure on specialized equipment and safety infrastructure. The high yields reported, often exceeding 90% for many substrates, translate directly into improved material throughput and reduced waste disposal costs, enhancing the overall economic viability of the process.
- Cost Reduction in Manufacturing: The economic argument for this process is compelling due to the substitution of costly coupling reagents and anhydrides with inexpensive, commercially available starting materials. By utilizing TFBen as a solid CO source, the process avoids the logistical and safety costs associated with storing and handling high-pressure carbon monoxide cylinders. Additionally, the high catalytic efficiency means that precious metal loading can be kept low while maintaining high turnover numbers, further driving down the cost per kilogram of the final product. The simplified workup procedure, which avoids complex extractions or recrystallizations in favor of direct chromatography or filtration, reduces solvent consumption and labor hours, contributing to substantial operational cost savings.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of widely available feedstocks that are not subject to the same supply constraints as specialized fluorinating agents or exotic heterocycles. The starting amines and the trifluoroethylimidoyl chloride precursor are produced on a large industrial scale, ensuring a stable and continuous supply even during market fluctuations. The robustness of the reaction conditions, which tolerate a broad range of functional groups, means that the process is less sensitive to minor variations in raw material quality, reducing the risk of batch failures. This reliability allows for more accurate production planning and shorter lead times for delivering high-purity intermediates to downstream customers, securing the continuity of the drug development timeline.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this method aligns well with modern green chemistry principles. The use of a solid CO surrogate minimizes the release of volatile organic compounds and toxic gases into the environment, simplifying compliance with increasingly stringent environmental regulations. The reaction has been demonstrated to be scalable, with the potential for expansion from gram-scale laboratory synthesis to multi-kilogram or ton-scale commercial production without significant loss of efficiency. The high atom economy of the cascade reaction ensures that a maximum proportion of the reactant mass is incorporated into the final product, minimizing the generation of chemical waste and reducing the burden on waste treatment facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the method's capabilities and limitations. Understanding these details is crucial for R&D teams evaluating this route for their specific project needs and for procurement teams assessing the feasibility of long-term supply agreements.
Q: What are the primary limitations of conventional quinazolinone synthesis methods?
A: Traditional methods often rely on harsh reaction conditions, expensive pre-activated substrates like trifluoroacetic anhydride, or coupling reagents like T3P, resulting in lower yields and narrow substrate compatibility.
Q: What serves as the carbon monoxide source in this novel protocol?
A: The method utilizes TFBen (1,3,5-tricarboxylic acid phenol ester) as a safe and effective carbon monoxide substitute, which releases CO in situ under heating conditions to drive the carbonylation.
Q: Has this method been validated for complex drug molecule synthesis?
A: Yes, the protocol was successfully applied to the total synthesis of the bioactive alkaloid Rutaecarpine, achieving a high overall yield of 77% across three steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing advanced synthetic technologies to accelerate drug discovery and development. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into reliable commercial supply. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required by global regulatory bodies. We are uniquely positioned to leverage the palladium-catalyzed carbonylation technology described in CN113045503B to deliver high-quality 2-trifluoromethyl quinazolinone intermediates that meet the exacting standards of the pharmaceutical industry.
We invite you to collaborate with us to explore how this innovative synthesis can optimize your specific project requirements. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your target molecule, demonstrating exactly how this route can improve your margins and timelines. We encourage you to contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your custom synthesis projects. Let us be your partner in turning complex chemical challenges into commercial successes.