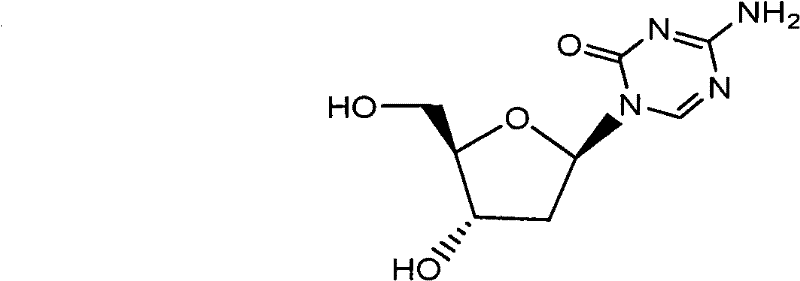
Advanced Decitabine Manufacturing: Overcoming Purification Bottlenecks for Commercial Scale
The pharmaceutical landscape for oncology treatments continues to evolve, with Decitabine standing out as a critical therapeutic agent for myelodysplastic syndromes (MDS). As detailed in patent CN101560233B, a novel preparation method has emerged that addresses long-standing inefficiencies in the synthesis of this 2'-deoxycytidine analogue. This technical insight report analyzes the strategic shift from traditional 2-deoxy-D-ribose starting materials to 1-methyl-2-deoxy-D-ribose, a modification that fundamentally alters the impurity profile and commercial viability of the process. For R&D Directors and Procurement Managers, understanding this mechanistic pivot is essential for securing a reliable API intermediate supplier capable of delivering high-purity materials without the prohibitive costs associated with legacy purification techniques.
The core innovation lies in the structural management of the sugar moiety during the initial acetylation phase. Conventional routes often struggle with the formation of stable byproducts that co-elute with the desired intermediate, necessitating repeated chromatographic separations that destroy yield. By introducing a methyl group at the 1-position of the ribose ring prior to acetylation, the new pathway effectively blocks the formation of the hexa-atomic nucleoside byproduct, historically identified as Formula VIII in prior literature. This structural safeguard ensures that the subsequent glycosylation step proceeds with higher fidelity, laying the groundwork for a process that is not only chemically superior but also economically robust for large-scale manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
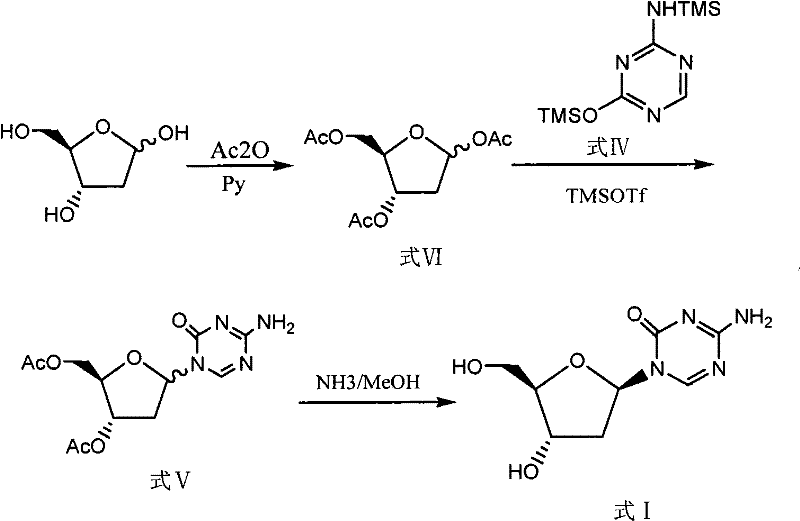
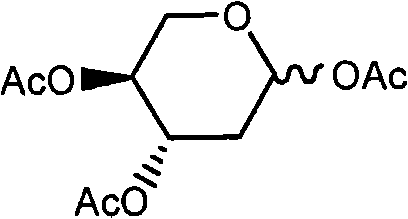
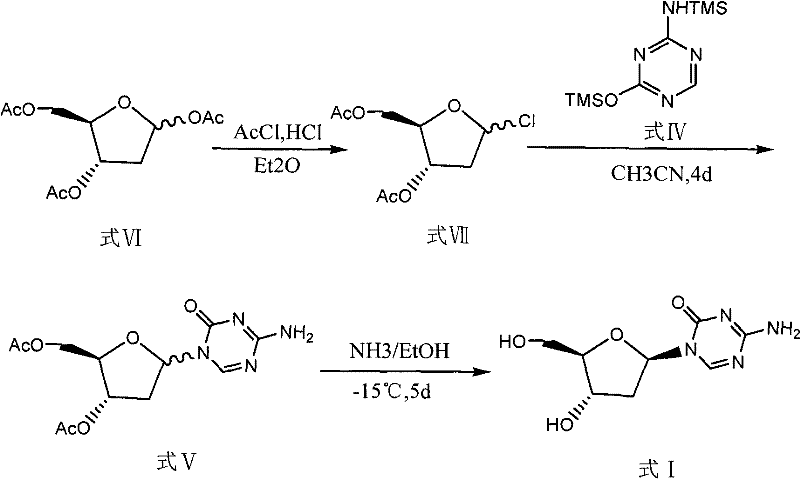
Historical methods for synthesizing Decitabine have been plagued by low overall yields and operational complexities that hinder commercial scale-up. One prominent prior art route involves the direct esterification of 2-deoxy-D-ribose, which frequently results in the generation of the hexa-atomic nucleoside byproduct (Formula VIII) in significant quantities, often ranging between 30% to 40% of the reaction mixture. This byproduct is chemically similar to the desired intermediate, making its removal via standard purification techniques exceptionally difficult and costly. Furthermore, alternative pathways utilizing hydrogen chloride gas in ether solvents to generate alpha-chloro intermediates suffer from extended reaction times, often requiring up to four days for the glycosylation step alone, followed by another five days for alcoholysis.
The reliance on column chromatography for purification in these legacy methods represents a critical bottleneck for industrial application. Column chromatography is inherently batch-limited, solvent-intensive, and difficult to automate, rendering it unsuitable for the production of metric ton quantities required by the global pharmaceutical market. The cumulative effect of these inefficiencies is a total recovery rate that frequently falls below 5%, driving up the cost of goods sold (COGS) to levels that are unsustainable for generic drug manufacturers. Additionally, the use of harsh reagents like hydrogen chloride gas introduces significant safety and environmental compliance challenges, further complicating the supply chain for high-purity pharmaceutical intermediates.
The Novel Approach
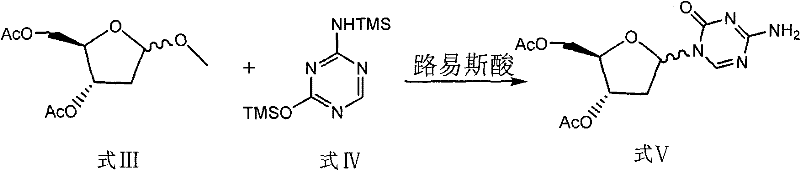
The methodology disclosed in CN101560233B offers a transformative solution by re-engineering the starting material strategy. By utilizing 1-methyl-2-deoxy-D-ribose (Formula II) as the initial raw material, the process circumvents the formation of the problematic hexa-atomic byproduct entirely. The acetylation of this methylated sugar proceeds under moderate conditions (0-30°C) with pyridine and acetic anhydride, yielding the 1-methyl-3,5-diacetoxy-2-deoxy-D-ribose (Formula III) with high selectivity. This intermediate then undergoes glycosylation with 2,4-bis-(trimethylsilyl)-5-azacytosine (Formula IV) in the presence of a Lewis acid catalyst, such as anhydrous stannic chloride or TMSOTf. The reaction is significantly faster and cleaner, occurring within 5 to 18 hours at temperatures between 0°C and 25°C.
Perhaps the most significant commercial advantage of this novel approach is the replacement of column chromatography with D315 macroporous weakly basic anion exchange resin for the final hydrolysis step. This resin-mediated deprotection allows for the removal of acetyl groups under mild conditions at room temperature within 2 to 6 hours. The use of a solid-phase resin simplifies the workup procedure to a filtration step, eliminating the need for large volumes of organic solvents and the labor associated with column packing and elution. This shift not only enhances the total recovery rate to approximately 27.4% but also aligns the manufacturing process with green chemistry principles, reducing waste generation and improving the overall environmental footprint of the production facility.
Mechanistic Insights into Lewis Acid-Catalyzed Glycosylation
The success of this synthesis hinges on the precise control of stereochemistry during the glycosylation step, where the sugar moiety is attached to the nucleobase. In the novel pathway, the Lewis acid catalyst activates the anomeric center of the diacetoxy sugar intermediate (Formula III), facilitating the nucleophilic attack by the silylated 5-azacytosine (Formula IV). The presence of the 1-methyl group on the sugar ring plays a crucial steric and electronic role, stabilizing the oxocarbenium ion intermediate and directing the attack to favor the desired beta-anomer configuration. This mechanistic control is vital for ensuring the biological activity of the final Decitabine product, as the alpha-anomer is inactive and constitutes a critical impurity that must be minimized. The use of solvents like 1,2-dichloroethane or methylene dichloride further optimizes the solubility of the reactants and the stability of the catalytic complex.
Impurity control is further enhanced during the final deprotection phase through the specific selection of the D315 resin. Unlike traditional alkaline hydrolysis which can lead to degradation of the sensitive 5-aza ring, the weakly basic anion exchange resin provides a buffered environment that selectively cleaves the ester bonds without compromising the integrity of the nucleoside structure. This selectivity is paramount for achieving the stringent purity specifications required for pharmaceutical applications, typically exceeding 99% purity with minimal alpha-anomer content. The resin's macroporous structure allows for efficient mass transfer, ensuring that the hydrolysis proceeds uniformly throughout the reaction mixture, thereby preventing the formation of partially deprotected intermediates that could complicate downstream crystallization.
How to Synthesize Decitabine Efficiently
The synthesis of Decitabine via this optimized route involves three distinct operational stages that can be seamlessly integrated into a standard multipurpose chemical plant. The process begins with the protection of the ribose starting material, followed by the critical coupling reaction, and concludes with the resin-mediated deprotection. Each step has been engineered to minimize isolation requirements and maximize throughput, making it an ideal candidate for continuous or semi-continuous processing. For technical teams looking to implement this route, the key lies in maintaining strict control over moisture levels during the glycosylation step and optimizing the resin loading ratio during hydrolysis to ensure complete conversion. Detailed standardized synthesis steps see the guide below.
- Acetylation of 1-methyl-2-deoxy-D-ribose with acetic anhydride in pyridine to form the diacetoxy intermediate.
- Lewis acid catalyzed glycosylation with 2,4-bis-(trimethylsilyl)-5-azacytosine to form the protected nucleoside.
- Hydrolysis using D315 macroporous weakly basic anion exchange resin to remove protecting groups and yield final Decitabine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthesis route offers tangible benefits that extend beyond simple yield improvements. The elimination of column chromatography represents a drastic reduction in operational expenditure, as it removes the need for expensive silica gel, vast quantities of high-purity solvents, and the significant labor hours required for manual purification. This simplification of the downstream processing allows for a more predictable production schedule, reducing the risk of batch failures and delays that often plague complex nucleoside manufacturing. Consequently, this leads to substantial cost savings in pharmaceutical intermediates manufacturing, making the final API more competitive in the global market.
- Cost Reduction in Manufacturing: The removal of column chromatography and the reduction in reaction times directly translate to lower utility and material costs. By avoiding the use of hazardous hydrogen chloride gas and replacing it with safer Lewis acid catalysts that can be quenched and recovered, the process reduces waste disposal costs and safety compliance burdens. The higher overall yield means that less raw material is required to produce the same amount of final product, significantly optimizing the cost of goods sold without compromising on quality standards.
- Enhanced Supply Chain Reliability: The starting materials for this process, specifically 1-methyl-2-deoxy-D-ribose, are commercially available and stable, reducing the risk of raw material shortages. The robustness of the resin-mediated hydrolysis step ensures that production is less susceptible to variations in operator skill or environmental conditions, leading to more consistent batch-to-batch quality. This reliability is crucial for reducing lead time for high-purity pharmaceutical intermediates, ensuring that downstream API manufacturers can maintain their own production schedules without interruption.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up of complex pharmaceutical intermediates in mind, utilizing unit operations that are easily transferable from pilot plant to full-scale production. The reduction in solvent usage and the elimination of toxic byproducts align with increasingly stringent environmental regulations, minimizing the need for expensive effluent treatment. This environmental compatibility ensures long-term operational continuity and reduces the risk of regulatory shutdowns, securing the supply chain against future compliance challenges.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Decitabine synthesis method. These insights are derived directly from the patent data and are intended to clarify the feasibility of adopting this route for large-scale production. Understanding these nuances is critical for stakeholders evaluating the potential for technology transfer or contract manufacturing partnerships.
Q: Why is 1-methyl-2-deoxy-D-ribose preferred over 2-deoxy-D-ribose in this synthesis?
A: Using 1-methyl-2-deoxy-D-ribose prevents the formation of the difficult-to-remove hexa-atomic nucleoside byproduct (Formula VIII) often seen in conventional esterification methods, significantly simplifying purification.
Q: How does the D315 resin method improve scalability compared to column chromatography?
A: The D315 macroporous weakly basic anion exchange resin allows for hydrolysis and purification in a single operational step without the need for labor-intensive and solvent-heavy column chromatography, making it viable for ton-scale production.
Q: What is the reported total yield advantage of this patent method?
A: The patent reports a total yield of approximately 27.4%, which is a substantial improvement over prior art methods that often yielded less than 5% due to complex purification losses.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Decitabine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for oncology therapeutics like Decitabine. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical advantages of this patent are fully realized in practice. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch of Decitabine intermediate meets the highest global pharmacopoeia standards, providing our partners with the confidence needed to advance their drug development pipelines.
We invite pharmaceutical companies and generic manufacturers to collaborate with us on optimizing their supply chains for nucleoside analogues. By leveraging our expertise in this novel resin-mediated hydrolysis technology, we can help you achieve significant efficiency gains and cost reductions. Please contact our technical procurement team to request a Customized Cost-Saving Analysis, where we will provide specific COA data and route feasibility assessments tailored to your volume requirements and quality targets.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →