Revolutionizing Decitabine Production: A Scalable Route for High-Purity Nucleoside Intermediates
Revolutionizing Decitabine Production: A Scalable Route for High-Purity Nucleoside Intermediates
The pharmaceutical industry constantly seeks robust synthetic pathways for oncology therapeutics, particularly for nucleoside analogs like Decitabine, a potent DNA methyltransferase inhibitor approved for myelodysplastic syndromes. A significant breakthrough in this domain is detailed in patent CN111471077B, which discloses a novel 2-deoxy-D-ribose derivative and its application in the stereoselective synthesis of Decitabine. This technology addresses a longstanding bottleneck in nucleoside chemistry: the difficulty in controlling the anomeric configuration during glycosidic bond formation. By introducing a specific mesylate leaving group on an Fmoc-protected sugar scaffold, the inventors have achieved a dramatic improvement in beta-isomer selectivity, a critical quality attribute for biological activity. For R&D directors and procurement specialists, this represents a pivotal shift towards more efficient, cost-effective, and reliable supply chains for high-value anticancer agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Decitabine has been plagued by poor stereocontrol, resulting in mixtures of alpha and beta anomers that are notoriously difficult to separate. Prior art methods, such as those reported by Piskala or using chlorodeoxyribose precursors, often rely on leaving groups like acetyl or chlorine which lack the necessary reactivity or steric influence to dictate high beta-selectivity. For instance, earlier routes frequently resulted in alpha:beta ratios hovering around 1:1 or even favoring the inactive alpha-isomer, necessitating extensive and yield-depleting recrystallization processes. Furthermore, the use of unstable chloro-sugars introduces safety hazards and storage complexities, while acetyl-protected intermediates often suffer from migration issues or insufficient activation. These inefficiencies translate directly into higher manufacturing costs, extended lead times, and inconsistent supply availability for downstream API producers.
The Novel Approach
The methodology outlined in CN111471077B circumvents these historical deficiencies through a cleverly designed intermediate, the 2-deoxy-D-ribose derivative (III). This approach utilizes 9-fluorenylmethoxycarbonyl (Fmoc) groups to protect the 3 and 5 hydroxyl positions, creating a bulky steric environment that inherently favors the formation of the beta-anomer during the subsequent coupling reaction. Crucially, the conversion of the C-1 methoxy group into a mesylate (methanesulfonate) ester creates a superior leaving group compared to traditional halides or acetates. This combination of steric bulk and enhanced leaving group ability allows the coupling with 5-azacytosine to proceed with exceptional stereoselectivity.
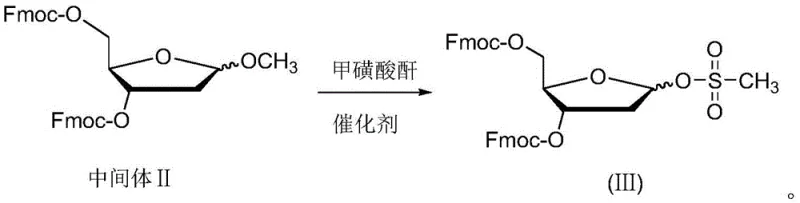
As illustrated in the reaction scheme, the transformation of Intermediate II into derivative (III) is achieved under mild acidic catalysis at low temperatures, ensuring high purity and yield. This strategic modification of the sugar moiety is the cornerstone of the invention, enabling a beta-to-alpha ratio of approximately 8:1 in the final coupling step, a substantial improvement over the 1:1 ratios seen in competing technologies. This leap in selectivity drastically simplifies downstream purification, making the process economically viable for large-scale commercialization.
Mechanistic Insights into Stereoselective Glycosylation
The success of this synthetic route lies in the intricate interplay between the protecting group strategy and the nature of the leaving group. In the coupling reaction (Step e in the full synthesis), the 2-deoxy-D-ribose derivative (III) reacts with hexamethyldisilazane-activated 5-azacytosine. The Fmoc groups at the 3 and 5 positions are significantly larger than the acetyl or benzoyl groups used in prior art. During the formation of the oxocarbenium ion intermediate, these bulky groups exert a profound steric influence, effectively blocking the alpha-face of the sugar ring. Consequently, the nucleophilic attack by the silylated base is directed almost exclusively from the beta-face. Additionally, the mesylate group at the C-1 position is an excellent leaving group, facilitating rapid ionization under Lewis acid catalysis (such as TMSOTf or SnCl4) without requiring harsh conditions that might degrade the sensitive triazine ring of the base.
Impurity control is another critical aspect where this mechanism excels. In conventional syntheses, the presence of alpha-anomers and their subsequent deprotection products creates a complex impurity profile that is challenging to purge. By shifting the thermodynamic and kinetic balance heavily towards the beta-isomer (8:1 ratio), the generation of alpha-related impurities is minimized at the source. This "quality by design" approach reduces the burden on purification units, such as chromatography or repeated crystallizations, which are often the most costly and time-consuming stages of API manufacturing. The result is a cleaner crude product with higher HPLC purity, streamlining the path to the final drug substance.
How to Synthesize 2-deoxy-D-ribose Derivative Efficiently
The preparation of the key intermediate (III) is a straightforward three-step sequence starting from commercially available 2-deoxy-D-ribose, designed for operational simplicity and high throughput. The process begins with the oxygen methylation of the anomeric hydroxyl group to form a stable methyl glycoside (Intermediate I), followed by the selective protection of the primary and secondary hydroxyls at positions 3 and 5 using Fmoc-Cl. The final and most critical step involves the sulfonation of the C-1 methoxy group using methanesulfonic anhydride. Detailed standardized operating procedures for temperature control, reagent addition rates, and workup protocols are essential to maintain the high stereointegrity and yield reported in the patent examples.
- Perform oxygen methylation of 2-deoxy-D-ribose using strong acid methanol solution to generate Intermediate I.
- Protect the 3,5-hydroxyl groups of Intermediate I with Fmoc-Cl under alkaline conditions to form Intermediate II.
- React Intermediate II with methanesulfonic anhydride and a catalyst to sulfonate the C-1 oxymethyl group, yielding the key derivative (III).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits beyond mere technical elegance. The primary advantage is the significant reduction in manufacturing costs driven by improved material efficiency. Because the reaction yields for the key intermediate (III) consistently exceed 90%, there is minimal waste of starting materials. Furthermore, the high stereoselectivity eliminates the need for expensive and yield-loss-prone separation of alpha and beta isomers, which traditionally consumes a large portion of the production budget. The elimination of complex purification steps translates directly into lower operational expenditures and a more competitive price point for the final nucleoside intermediate.
- Cost Reduction in Manufacturing: The process utilizes readily available reagents such as methanesulfonic anhydride and Fmoc-Cl, avoiding the need for exotic or prohibitively expensive catalysts. The high yield of the sulfonation step (>90%) ensures that raw material costs are optimized. Moreover, by preventing the formation of difficult-to-remove alpha-isomers, the process saves substantial costs associated with solvent usage, energy consumption for recrystallization, and labor hours for purification. This efficiency makes the cost reduction in nucleoside intermediate manufacturing substantial and sustainable.
- Enhanced Supply Chain Reliability: The robustness of this chemistry enhances supply security. The reactions are performed under mild conditions (typically between -20°C and 10°C), which can be easily maintained using standard industrial cooling systems rather than specialized cryogenic equipment. This accessibility reduces the risk of batch failures due to equipment limitations. Additionally, the intermediates generated, particularly the Fmoc-protected species, exhibit good stability, allowing for safer storage and transportation between different manufacturing sites if a multi-vendor strategy is employed.
- Scalability and Environmental Compliance: The patent examples demonstrate successful execution on a kilogram scale (e.g., using 5000ml reactors), indicating readiness for commercial scale-up of complex nucleoside intermediates. The use of common organic solvents like chloroform and acetonitrile simplifies solvent recovery and recycling programs, aiding in environmental compliance. The absence of heavy metal catalysts in the initial steps (using acid catalysis instead) further simplifies waste treatment and reduces the environmental footprint of the manufacturing process.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and beneficial effects described in CN111471077B, providing a clear picture of what partners can expect when integrating this route into their production pipelines. Understanding these nuances is vital for making informed sourcing and development decisions.
Q: How does this new derivative improve beta-isomer selectivity compared to traditional methods?
A: Traditional methods often yield alpha:beta ratios near 1:1 or 3:2. This patent utilizes bulky Fmoc protecting groups at the 3 and 5 positions combined with a highly reactive mesylate leaving group at C-1. This specific steric and electronic environment directs the nucleophilic attack of 5-azacytosine to favor the beta-configuration, achieving a superior ratio of approximately 8:1.
Q: What are the yield advantages of synthesizing the 2-deoxy-D-ribose derivative (III)?
A: The preparation of the key intermediate (III) is highly efficient, with reported yields exceeding 90% in multiple examples. The process avoids complex purification steps typically required for separating alpha and beta anomers early in the synthesis, significantly reducing material loss.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method is suitable for industrial production. It operates under mild conditions (often between -20°C and 10°C for critical steps), uses common solvents like chloroform and acetonitrile, and does not require specialized high-pressure or cryogenic equipment, facilitating easy scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Decitabine Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic route described in CN111471077B for the global oncology market. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high stereoselectivity and yields demonstrated in the lab are faithfully reproduced at an industrial level. Our rigorous QC labs and stringent purity specifications guarantee that every batch of 2-deoxy-D-ribose derivative meets the exacting standards required for GMP API synthesis, providing our clients with peace of mind and regulatory confidence.
We invite forward-thinking pharmaceutical companies to collaborate with us to optimize their Decitabine supply chain. By leveraging this advanced chemistry, we can offer a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can accelerate your time-to-market for this critical anticancer therapy.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →