Advanced Synthetic Routes for High-Purity Capecitabine Intermediates and Commercial Scalability
Advanced Synthetic Routes for High-Purity Capecitabine Intermediates and Commercial Scalability
The pharmaceutical industry continuously seeks robust synthetic pathways for oncology therapeutics, particularly for oral prodrugs like Capecitabine. Patent CN101469008A introduces a transformative approach to synthesizing Capecitabine hydroxy derivatives, addressing critical bottlenecks in the final deprotection stages of traditional manufacturing. This technology leverages a specialized orthoester protecting group strategy that fundamentally alters the reaction landscape, allowing for the removal of protecting groups under significantly milder acidic or alkaline conditions compared to historical methods. By shifting away from harsh basic hydrolysis, this innovation ensures that the sensitive fluoropyrimidine core remains intact, thereby drastically reducing the formation of degradation byproducts and impurities. For R&D directors and process chemists, this represents a pivotal advancement in achieving high-purity intermediates that streamline the path to commercial API production while maintaining rigorous quality standards.
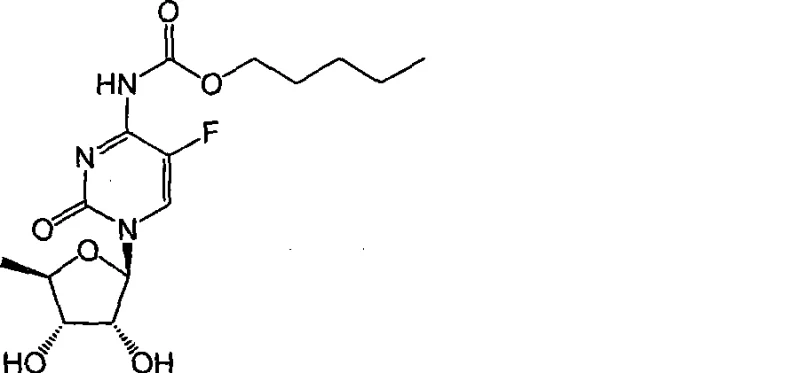
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Capecitabine has been plagued by the reliance on aggressive reaction conditions during the final deprotection steps. As detailed in the background art of the patent, conventional routes (such as those utilizing triacetoxy ribofuranose or direct acylation of 5'-deoxy-5-fluoro-cytidine) typically necessitate strong basic environments to cleave hydroxyl protecting groups. These harsh alkaline conditions are inherently problematic because they lack selectivity, often triggering unwanted side reactions that compromise the structural integrity of the nucleoside analogue. The result is a crude product with low purity and a complex impurity profile that is notoriously difficult to purify, often requiring extensive chromatographic separation which is economically unviable at scale. Furthermore, the poor controllability of these exothermic basic hydrolysis reactions poses significant safety risks and batch-to-batch consistency issues, making it challenging for supply chain managers to guarantee reliable delivery of high-quality material.
The Novel Approach
In stark contrast, the novel methodology disclosed in CN101469008A employs a sophisticated orthoester protection strategy that circumvents the need for destructive basic conditions. By introducing a specific protecting group derived from tetraalkyl orthocarbonates, the synthesis creates an intermediate (Formula III) that is stable during the acylation phase but highly labile under mild hydrolytic conditions. This allows the final deprotection to proceed in weakly acidic or alkaline media, preserving the stereochemistry and chemical stability of the molecule. The process controllability is markedly enhanced, leading to a crude product of such high purity that it can meet United States Pharmacopoeia (USP) standards without the need for繁琐 (tedious) purification treatments. This shift not only simplifies the downstream processing but also significantly boosts the overall yield and economic efficiency of the manufacturing process, offering a compelling value proposition for large-scale API production.
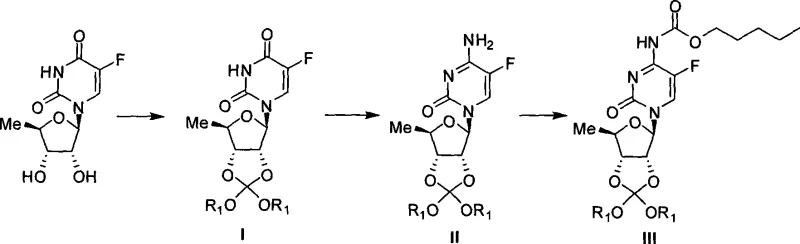
Mechanistic Insights into Orthoester Protection and Mild Hydrolysis
The core mechanistic advantage of this technology lies in the unique stability profile of the orthoester functionality installed at the 2',3'-positions of the ribose ring. In the initial protection step, 5'-deoxy-5-fluoro-uridine or cytidine reacts with tetraalkyl orthocarbonate in the presence of an acidic catalyst, such as p-toluenesulfonic acid or boron trifluoride, to form a cyclic orthoester. This structure effectively masks the reactive hydroxyl groups, preventing them from interfering during the subsequent N-acylation of the pyrimidine base. Unlike simple acetate esters which require strong bases for cleavage, the orthoester linkage is susceptible to hydrolysis under much gentler conditions. The mechanism involves protonation of the orthoester oxygen followed by nucleophilic attack by water, which proceeds rapidly at controlled pH levels between 1 and 6, or alternatively under mild alkaline conditions (pH 7-11). This precise tunability allows process chemists to optimize the reaction environment to maximize the rate of deprotection while minimizing the risk of hydrolyzing the carbamate linkage at the N1 position or degrading the fluorinated base.
From an impurity control perspective, this mild hydrolysis mechanism is paramount for ensuring the quality of the final Active Pharmaceutical Ingredient (API). Traditional strong base hydrolysis often leads to epimerization at the anomeric center or degradation of the urethane linkage, generating structurally related impurities that are difficult to separate. By operating within a narrow, mild pH window, the novel process suppresses these degradation pathways, resulting in a cleaner reaction profile. The patent data indicates that the crude product obtained directly from this hydrolysis step possesses sufficient purity to meet stringent regulatory specifications, effectively eliminating the need for resource-intensive recrystallization or column chromatography steps. This inherent purity is a direct consequence of the chemoselective nature of the orthoester deprotection, providing a robust safeguard against the formation of genotoxic or carcinogenic impurities that often arise from harsh chemical treatments in nucleoside synthesis.
How to Synthesize Capecitabine Efficiently
The synthesis of Capecitabine via this novel pathway is designed for operational simplicity and scalability, making it an ideal candidate for technology transfer from laboratory to pilot plant. The process generally follows a logical sequence starting from commercially available nucleoside precursors, utilizing standard organic synthesis unit operations that are familiar to most contract development and manufacturing organizations (CDMOs). The key to success lies in the precise control of reaction parameters during the protection and acylation stages to ensure the formation of the correct regioisomer before the final hydrolytic release. Detailed standard operating procedures regarding stoichiometry, solvent selection, and temperature profiles are critical for maximizing yield and minimizing waste. For a comprehensive breakdown of the specific experimental conditions and workup procedures required to execute this synthesis effectively, please refer to the standardized guide below.
- Protect the 2',3'-diol of 5'-deoxy-5-fluoro-cytidine using tetraalkyl orthocarbonate under acidic catalysis to form the stable orthoester intermediate.
- Perform selective N-acylation on the amino group using activated carbonate reagents like n-pentyl chloroformate in aprotic solvents.
- Execute the final deprotection via mild acidic or alkaline hydrolysis to remove the orthoester group without degrading the sensitive fluoropyrimidine core.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this orthoester-based synthetic route offers tangible economic and logistical benefits that extend far beyond simple yield improvements. By fundamentally altering the chemistry of the final deprotection step, the process eliminates the need for expensive and hazardous strong base reagents, replacing them with milder, more manageable alternatives. This shift reduces the burden on waste treatment facilities and lowers the overall cost of goods sold (COGS) by minimizing solvent consumption and energy usage associated with extensive purification workflows. Furthermore, the high controllability of the reaction ensures consistent batch quality, reducing the risk of production delays caused by out-of-specification results. This reliability is crucial for maintaining a steady supply of critical oncology medications in a volatile global market, allowing manufacturers to plan inventory with greater confidence and reduce safety stock requirements.
- Cost Reduction in Manufacturing: The elimination of complex purification steps, such as preparative chromatography, translates directly into substantial cost savings. Since the crude product meets USP standards immediately after hydrolysis, manufacturers can bypass expensive resin columns and reduce solvent recovery costs. Additionally, the use of milder reagents reduces corrosion on reactor vessels and lowers the safety infrastructure costs associated with handling hazardous strong bases. The overall reduction in processing time and material waste contributes to a leaner, more cost-effective manufacturing model that enhances profit margins without compromising product quality.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures a more predictable production schedule. Because the reaction conditions are less sensitive to minor fluctuations in temperature or reagent quality compared to harsh basic hydrolysis, the risk of batch failure is significantly mitigated. This stability allows for tighter production planning and shorter lead times, ensuring that downstream formulation teams receive their API intermediates on schedule. Moreover, the starting materials, such as 5'-deoxy-5-fluoro-cytidine and tetraalkyl orthocarbonates, are readily available from multiple global suppliers, reducing the risk of raw material shortages and strengthening the resilience of the supply chain against geopolitical disruptions.
- Scalability and Environmental Compliance: Scaling chemical processes often amplifies safety and environmental challenges, but this mild hydrolysis method is inherently safer for large-scale operations. The absence of exothermic strong base reactions reduces the thermal load on reactors, simplifying heat management and cooling requirements. From an environmental standpoint, the reduction in solvent waste and the avoidance of hazardous reagents align with green chemistry principles, facilitating easier compliance with increasingly strict environmental regulations. This makes the technology not only commercially viable but also sustainable, appealing to stakeholders who prioritize environmental, social, and governance (ESG) criteria in their supplier selection process.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this orthoester protection technology in Capecitabine manufacturing. These insights are derived directly from the experimental data and comparative analysis presented in the patent literature, providing a factual basis for decision-making. Understanding these nuances is essential for R&D teams evaluating process changes and procurement officers assessing supplier capabilities. The answers highlight the specific advantages of this method over legacy technologies, focusing on purity, safety, and operational efficiency.
Q: Why is the orthoester protection strategy superior to conventional acetyl protection for Capecitabine?
A: Conventional methods often require strong basic conditions for final deprotection, which induces side reactions and lowers crude purity. The orthoester strategy allows for removal under weakly acidic or alkaline conditions, significantly enhancing process controllability and final product quality.
Q: What are the critical starting materials for this synthetic route?
A: The process utilizes readily available precursors such as 5'-deoxy-5-fluoro-cytidine or 5'-deoxy-5-fluoro-uridine, along with tetraalkyl orthocarbonates for protection and activated carbonates like n-pentyl chloroformate for the acylation step.
Q: Does this method meet international pharmacopoeia standards?
A: Yes, the patent explicitly states that the crude product obtained through this mild hydrolysis method possesses high purity that meets United States Pharmacopoeia (USP) standards without requiring complex purification treatments.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Capecitabine Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic technologies to maintain competitiveness in the global pharmaceutical market. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovations like the orthoester protection strategy described in CN101469008A can be seamlessly translated into industrial reality. We are committed to delivering high-purity Capecitabine intermediates that adhere to stringent purity specifications, leveraging our rigorous QC labs to verify every batch against international pharmacopoeia standards. Our state-of-the-art facilities are equipped to handle the specific solvent systems and mild reaction conditions required for this chemistry, guaranteeing a supply of material that supports your clinical and commercial needs with unwavering consistency.
We invite you to collaborate with us to optimize your supply chain and reduce manufacturing costs through the adoption of this superior synthetic route. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this technology can improve your bottom line. Please contact us today to request specific COA data for our Capecitabine intermediates and to discuss route feasibility assessments for your upcoming projects. Let us partner with you to bring safer, more effective oncology treatments to patients worldwide through chemical excellence and supply chain reliability.