Advanced Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Commercial API Production
Advanced Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic methodologies to construct complex heterocyclic scaffolds efficiently, particularly those containing trifluoromethyl groups which enhance metabolic stability and lipophilicity. Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds, a structural motif prevalent in high-value drugs such as Sitagliptin, Maraviroc, and Deferasirox. This technology represents a significant leap forward by utilizing a non-metallic iodine-promoted cascade reaction that bypasses the need for expensive transition metal catalysts. For R&D directors and procurement specialists, this innovation offers a pathway to high-purity intermediates with reduced environmental impact and simplified purification protocols. The method leverages readily available aryl ethyl ketones and trifluoroethylimide hydrazides, ensuring a stable supply chain for critical API precursors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing polysubstituted 1,2,4-triazoles often rely heavily on transition metal catalysis, involving precious metals like palladium or copper which introduce significant cost burdens and regulatory hurdles regarding residual heavy metals in final drug substances. Furthermore, many existing protocols demand stringent reaction conditions, such as strictly anhydrous and oxygen-free environments, which necessitate specialized equipment and increase operational complexity in a manufacturing setting. These conventional methods frequently suffer from limited substrate scope, struggling to accommodate diverse functional groups without compromising yield or selectivity. Additionally, the multi-step nature of older syntheses often results in lower overall atom economy and generates substantial chemical waste, posing challenges for environmental compliance and cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
The novel methodology described in the patent overcomes these barriers through an elegant iodine-promoted tandem reaction sequence performed in dimethyl sulfoxide (DMSO). This approach initiates with an iodination and Kornblum oxidation of aryl ethyl ketones to generate reactive aryl diketone intermediates in situ, which subsequently undergo condensation and cyclization with trifluoroethylimide hydrazides. 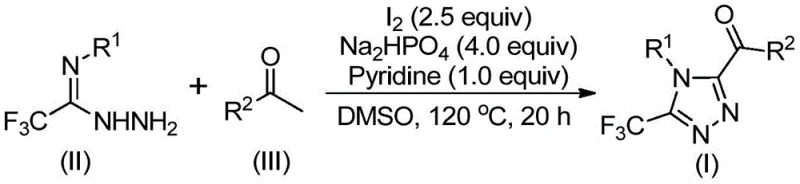 . This one-pot strategy eliminates the isolation of unstable intermediates and operates effectively under air without the need for inert gas protection. By replacing toxic heavy metals with inexpensive elemental iodine, the process drastically simplifies the workup procedure, typically requiring only filtration and standard chromatography, thereby enhancing the overall feasibility for commercial scale-up of complex pharmaceutical intermediates.
. This one-pot strategy eliminates the isolation of unstable intermediates and operates effectively under air without the need for inert gas protection. By replacing toxic heavy metals with inexpensive elemental iodine, the process drastically simplifies the workup procedure, typically requiring only filtration and standard chromatography, thereby enhancing the overall feasibility for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of oxidation and condensation steps driven by the unique reactivity of the iodine-DMSO system. Initially, elemental iodine facilitates the alpha-iodination of the aryl ethyl ketone, followed by a Kornblum oxidation where DMSO acts as the oxygen source to convert the methyl ketone into an alpha-dicarbonyl species. This highly electrophilic diketone intermediate then reacts with the nucleophilic nitrogen of the trifluoroethylimide hydrazide to form a hydrazone linkage. Under the continued influence of iodine and the basic environment provided by sodium dihydrogen phosphate and pyridine, an intramolecular cyclization occurs, closing the triazole ring and establishing the final 3,4,5-substitution pattern. This mechanism ensures high regioselectivity and minimizes the formation of isomeric byproducts, which is critical for maintaining the stringent purity specifications required in API synthesis.
From an impurity control perspective, the mild yet effective nature of this catalytic system allows for excellent functional group tolerance, accommodating substituents such as halogens, alkoxy groups, and trifluoromethyl groups on both the ketone and hydrazide components. 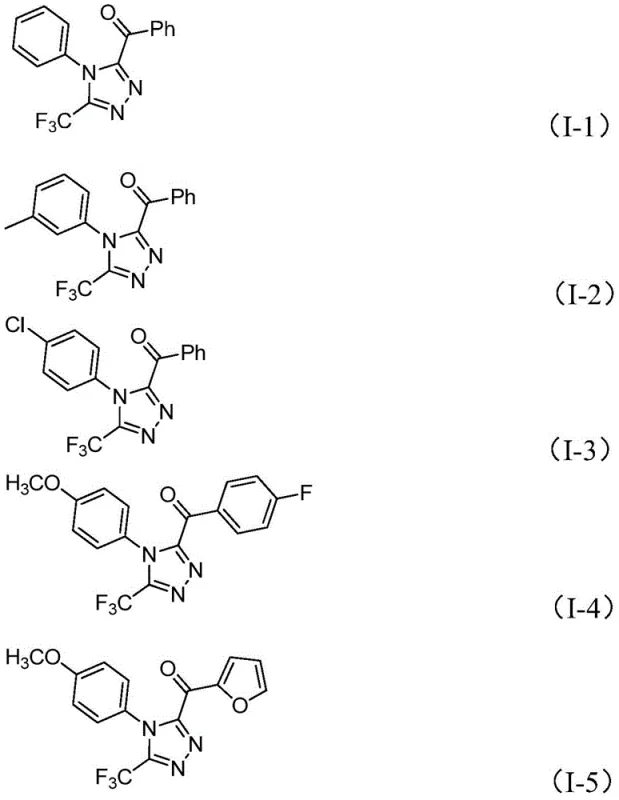 . The absence of aggressive reagents reduces the risk of decomposition or side reactions that often plague sensitive fluorinated compounds. Consequently, the resulting crude products exhibit cleaner profiles, reducing the burden on downstream purification units. This mechanistic robustness translates directly to higher batch-to-batch consistency, a key metric for supply chain reliability when producing high-purity pharmaceutical intermediates for global markets.
. The absence of aggressive reagents reduces the risk of decomposition or side reactions that often plague sensitive fluorinated compounds. Consequently, the resulting crude products exhibit cleaner profiles, reducing the burden on downstream purification units. This mechanistic robustness translates directly to higher batch-to-batch consistency, a key metric for supply chain reliability when producing high-purity pharmaceutical intermediates for global markets.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The operational simplicity of this synthesis makes it highly attractive for process chemists aiming to transfer laboratory protocols to pilot or production scales. The procedure involves a sequential addition of reagents where temperature control is the primary variable, shifting from an initial oxidation phase at 90-110°C to a cyclization phase at 110-130°C. Detailed standardized synthesis steps see the guide below, which outlines the precise molar ratios and timing required to maximize yield while minimizing reagent consumption. This streamlined workflow reduces the need for complex reactor configurations, allowing facilities to utilize standard glass-lined or stainless steel reactors commonly found in fine chemical manufacturing plants.
- Mix aryl ethyl ketone and elemental iodine in DMSO and heat to 90-110°C for 4-6 hours to initiate oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours to complete the cyclization, then purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis offers tangible strategic benefits centered around cost efficiency and supply continuity. The reliance on commodity chemicals such as acetophenones, elemental iodine, and DMSO ensures that raw material sourcing is not bottlenecked by exotic or geopolitically sensitive supply chains. The elimination of precious metal catalysts removes a major cost driver and mitigates the risk of price volatility associated with metals like palladium. Furthermore, the simplified post-treatment process reduces solvent consumption and waste disposal costs, contributing to a leaner manufacturing footprint. These factors collectively enable a more resilient supply chain capable of meeting the rigorous demands of the global pharmaceutical market.
- Cost Reduction in Manufacturing: The substitution of expensive transition metal catalysts with low-cost elemental iodine results in significant direct material savings, while the one-pot nature of the reaction reduces labor and energy costs associated with intermediate isolation and multiple reactor charges. The high atom economy of the cascade reaction ensures that a greater proportion of input mass is converted into valuable product, further driving down the cost per kilogram of the final intermediate. Additionally, the avoidance of specialized anhydrous conditions lowers infrastructure and utility expenses, making the process economically viable even for smaller batch sizes.
- Enhanced Supply Chain Reliability: Since the starting materials are widely available bulk chemicals with established global supply networks, the risk of production delays due to raw material shortages is minimized. The robustness of the reaction conditions, which do not require strict exclusion of moisture or oxygen, reduces the likelihood of batch failures caused by environmental fluctuations or equipment leaks. This operational stability ensures consistent output volumes, allowing manufacturers to honor long-term supply agreements with confidence and maintain steady inventory levels for their clients.
- Scalability and Environmental Compliance: The process has been demonstrated to scale easily from gram to multi-kilogram levels without loss of efficiency, facilitating a smooth transition from R&D to commercial production. The use of DMSO, a high-boiling polar aprotic solvent, allows for safe handling at elevated temperatures, while the absence of toxic heavy metals simplifies wastewater treatment and aligns with increasingly strict environmental regulations. This green chemistry profile enhances the corporate sustainability image of manufacturers and reduces the regulatory burden associated with hazardous waste disposal.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this triazole synthesis technology. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on reaction parameters and product quality. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the patented method utilizes elemental iodine as a non-metal promoter, eliminating the need for costly palladium or copper catalysts and simplifying heavy metal removal processes.
Q: What are the typical reaction conditions for this triazole formation?
A: The reaction proceeds in dimethyl sulfoxide (DMSO) at temperatures between 110°C and 130°C for 12 to 20 hours, without requiring strict anhydrous or oxygen-free environments.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the process uses cheap, commercially available raw materials and simple post-treatment steps like filtration and column chromatography, making it highly scalable for industrial applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic methodologies play in accelerating drug development and commercialization. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries like this iodine-promoted triazole synthesis can be successfully translated into industrial reality. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch of intermediate meets the exacting standards required by top-tier pharmaceutical companies. Our commitment to quality and technical excellence makes us a trusted partner for complex organic synthesis projects.
We invite you to collaborate with us to leverage this advanced technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our capabilities can optimize your supply chain and reduce your overall manufacturing costs.