Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
Introduction to Advanced Triazole Synthesis Technology
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for more efficient, cost-effective, and environmentally benign synthetic routes. A significant breakthrough in this domain is detailed in patent CN113105402B, which discloses a novel preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds. These heterocyclic scaffolds are critical structural motifs found in numerous high-value active pharmaceutical ingredients (APIs), including blockbuster drugs such as Maraviroc, Sitagliptin, and Deferasirox, as illustrated in the structural diversity shown below. The introduction of a trifluoromethyl group into these heterocycles is particularly valuable, as it significantly enhances physicochemical properties like metabolic stability, lipophilicity, and bioavailability, which are paramount for modern drug design.
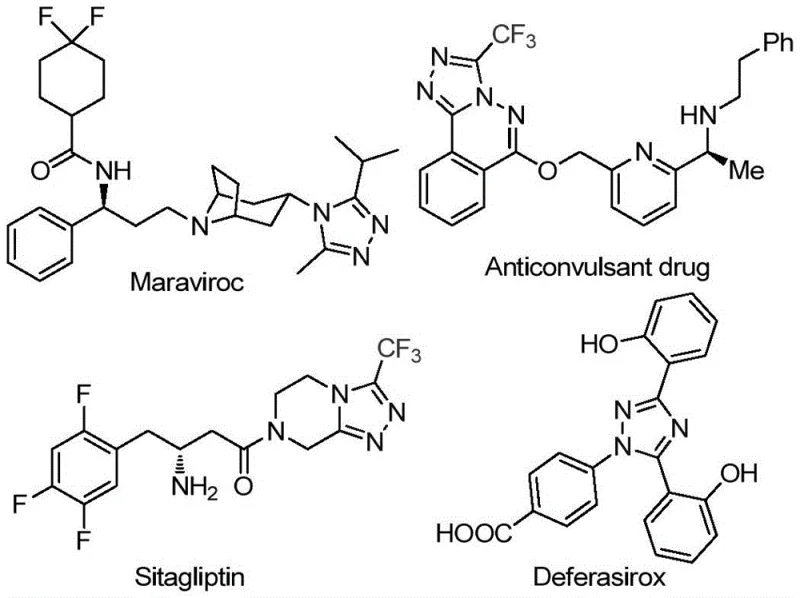
Despite the clear therapeutic potential of these molecules, traditional synthetic methodologies often suffer from significant limitations, including the use of toxic heavy metal catalysts, harsh reaction conditions, and complex multi-step sequences that hinder scalability. The technology described in CN113105402B addresses these pain points directly by offering a simple, efficient, and metal-free approach. By utilizing cheap and readily available starting materials like aryl ethanones and trifluoroethylimine hydrazides, this method provides a robust pathway for producing high-purity pharmaceutical intermediates. For R&D directors and procurement managers alike, understanding the nuances of this iodine-promoted cyclization is essential for optimizing supply chains and reducing the overall cost of goods sold (COGS) in API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles, particularly those bearing both trifluoromethyl and acyl groups, has been a challenging endeavor for organic chemists. Conventional routes frequently rely on transition metal catalysis, which introduces severe complications regarding residual metal removal—a critical quality attribute for pharmaceutical products that requires extensive and expensive purification steps. Furthermore, many existing protocols demand stringent anhydrous and oxygen-free environments, necessitating specialized equipment and increasing operational complexity. These factors collectively contribute to longer lead times and higher production costs, creating bottlenecks for reliable pharmaceutical intermediate suppliers aiming to meet the rigorous demands of global regulatory bodies. Additionally, the substrate scope in older methods is often narrow, limiting the ability to introduce diverse functional groups required for structure-activity relationship (SAR) studies.
The Novel Approach
In stark contrast, the methodology outlined in patent CN113105402B represents a paradigm shift towards greener and more practical synthesis. This novel approach leverages elemental iodine as a non-metal promoter in dimethyl sulfoxide (DMSO), effectively bypassing the need for toxic heavy metals. The reaction proceeds through a tandem sequence involving iodination and Kornblum oxidation, followed by cyclization, all within a single pot. This streamlined process not only simplifies the operational workflow but also dramatically reduces the environmental footprint by minimizing waste generation. The tolerance for various functional groups on both the aryl ketone and the hydrazide components allows for the rapid generation of diverse compound libraries, facilitating faster drug discovery cycles. By eliminating the need for inert atmosphere conditions, this method significantly lowers the barrier to entry for scale-up, making it an attractive option for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Iodine-Promoted Cyclization
To fully appreciate the technical robustness of this synthesis, one must delve into the mechanistic underpinnings that drive the formation of the 1,2,4-triazole core. The reaction initiates with the activation of the aryl ethyl ketone by iodine in DMSO, leading to an iodination event followed by a Kornblum oxidation to generate an aryl diketone intermediate in situ. This reactive species then undergoes a dehydration condensation with the trifluoroethylimine hydrazide to form a hydrazone intermediate. The final step involves an intramolecular cyclization promoted by the synergistic action of iodine and the base system (sodium dihydrogen phosphate and pyridine), closing the ring to yield the target 3,4,5-trisubstituted 1,2,4-triazole. This cascade mechanism is highly efficient, minimizing the accumulation of stable intermediates that could otherwise lead to impurity profiles difficult to control.
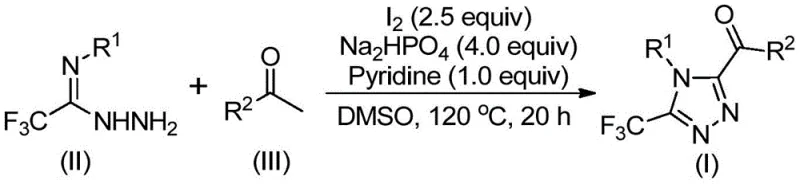
From an impurity control perspective, the choice of reagents and conditions plays a pivotal role. The use of sodium dihydrogen phosphate and pyridine creates a buffered environment that facilitates the cyclization while suppressing side reactions such as over-oxidation or polymerization. The specific molar ratios optimized in the patent—typically 1:2:4:1:2.5 for hydrazide, ketone, phosphate, pyridine, and iodine—ensure that the reaction kinetics favor the desired cyclization pathway. Furthermore, the moderate temperature range of 110-130°C is sufficient to drive the reaction to completion without inducing thermal degradation of the sensitive trifluoromethyl group. This precise control over reaction parameters ensures that the resulting high-purity pharmaceutical intermediates meet the stringent specifications required for downstream API synthesis, thereby reducing the risk of batch failures and ensuring consistent quality.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazoles Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to the sequential addition of reagents and temperature control to maximize yield and purity. The process is designed to be operationally simple, avoiding the complexities associated with air-sensitive chemistry. The following guide outlines the standardized protocol derived from the patent data, ensuring reproducibility and safety during execution. For detailed standard operating procedures and safety data sheets, please refer to the technical documentation provided by our engineering team.
- Mix aryl ethyl ketone and iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate oxidation.
- Add sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the mixture.
- Heat the reaction to 110-130°C for 12-20 hours, then filter and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis offers tangible strategic advantages that extend beyond mere chemical efficiency. The elimination of heavy metal catalysts translates directly into significant cost savings by removing the need for expensive scavengers and complex filtration systems typically required to meet residual metal limits. Moreover, the reliance on commodity chemicals such as aryl ethanones and elemental iodine ensures a stable and resilient supply chain, mitigating the risks associated with sourcing exotic or regulated reagents. This stability is crucial for maintaining continuous production schedules and avoiding disruptions that can ripple through the entire pharmaceutical value chain.
- Cost Reduction in Manufacturing: The economic benefits of this process are substantial, primarily driven by the simplification of the reaction setup and workup procedures. By operating without the need for anhydrous solvents or inert gas protection, facilities can utilize standard glass-lined reactors, significantly lowering capital expenditure requirements. The high atom economy and the use of inexpensive promoters like iodine further contribute to a lower cost per kilogram of the final intermediate. Additionally, the simplified purification process, often requiring only filtration and standard column chromatography, reduces solvent consumption and waste disposal costs, aligning with green chemistry principles and corporate sustainability goals.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply chain reliability by reducing the dependency on specialized reagents that may have long lead times or volatile pricing. Since the starting materials are widely available from multiple global suppliers, procurement teams can diversify their vendor base to negotiate better terms and ensure continuity of supply. The scalability of the method, demonstrated from gram to multi-kilogram levels in the patent examples, means that transitions from clinical trial material to commercial production can be executed smoothly without the need for extensive process re-engineering, thereby reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this method offers a cleaner profile that simplifies compliance with increasingly strict environmental regulations. The absence of heavy metals reduces the burden on wastewater treatment facilities and minimizes the generation of hazardous solid waste. The use of DMSO, a solvent with a relatively favorable safety profile compared to chlorinated alternatives, further supports environmental compliance efforts. The ability to easily scale this process to industrial quantities ensures that manufacturers can meet growing market demand for trifluoromethyl-containing APIs without compromising on environmental standards or operational safety.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent literature, providing a clear understanding of the method's capabilities and limitations for potential partners and stakeholders.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the patented method utilizes elemental iodine as a non-metal promoter, eliminating the need for costly heavy metal catalysts and simplifying purification.
Q: What are the optimal reaction conditions for high yield?
A: The preferred molar ratio is 1:2:4:1:2.5 for hydrazide, ketone, phosphate, pyridine, and iodine respectively, with a two-stage heating profile up to 130°C in DMSO.
Q: Is the process suitable for large-scale industrial production?
A: Yes, the method avoids strict anhydrous or oxygen-free conditions and uses cheap, commercially available raw materials, making it highly scalable for industrial application.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development and optimizing production costs. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to full-scale manufacturing is seamless and efficient. We are committed to delivering high-purity intermediates that adhere to stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch.
We invite you to collaborate with us to leverage this innovative iodine-promoted synthesis for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise can drive value and efficiency in your supply chain.