Advanced Triazolopyrimidine Synthesis for Potent USP28 Inhibitor Development
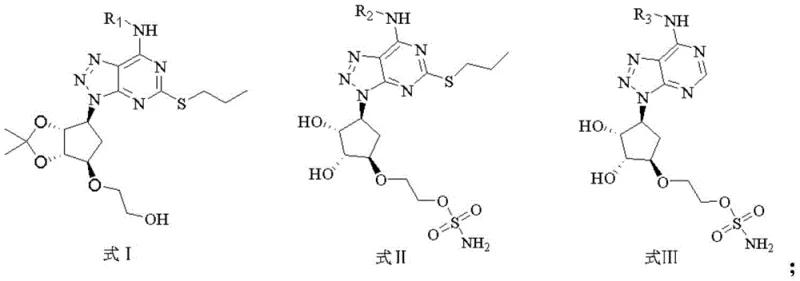
The pharmaceutical landscape is continuously evolving with the discovery of novel targets for oncology treatment, and the recent disclosure in patent CN114805367A marks a significant advancement in the field of deubiquitinating enzyme inhibitors. This intellectual property introduces a series of triazolopyrimidine derivatives characterized by general formulas I, II, and III, which demonstrate remarkable potency against USP28. For research and development teams focused on anti-tumor drug discovery, understanding the structural nuances and synthetic accessibility of these molecules is critical. The patent details a robust methodology for constructing these complex heterocyclic systems, providing a clear pathway from readily available starting materials to high-value biological probes. As a reliable pharmaceutical intermediate supplier, analyzing such patents allows us to bridge the gap between academic discovery and commercial viability, ensuring that promising candidates like these can be produced with the necessary purity and consistency for preclinical evaluation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fused triazole-pyrimidine systems has often been plagued by harsh reaction conditions and limited substrate scope, which can severely restrict the ability to perform structure-activity relationship (SAR) studies efficiently. Traditional routes frequently require extreme temperatures or hazardous reagents that complicate purification and lower overall yields, creating bottlenecks in the supply chain for medicinal chemists. Furthermore, older methodologies often lack the modularity needed to easily swap out substituents at key positions, such as the N-7 position or the C-5 sulfur linkage, which are crucial for optimizing binding affinity to the USP28 active site. These inefficiencies not only drive up the cost of goods but also extend the lead time for generating diverse libraries of analogues, slowing down the critical decision-making process in drug discovery pipelines.
The Novel Approach
The methodology outlined in CN114805367A presents a transformative solution by employing a modular synthetic strategy that allows for the systematic variation of R1, R2, and R3 groups while maintaining a stable core scaffold. This novel approach utilizes a condensation reaction between specific precursors followed by a controlled diazotization step, enabling the precise installation of diverse amine functionalities under relatively mild conditions. By leveraging common solvents such as ethanol and acetonitrile, the process minimizes environmental impact and simplifies downstream processing, making it highly attractive for cost reduction in pharmaceutical intermediate manufacturing. The ability to access three distinct structural families (Formulas I, II, and III) through slight modifications in the final steps provides researchers with a versatile toolkit for exploring the chemical space around the USP28 target without reinventing the wheel for each new analogue.
Mechanistic Insights into Triazolopyrimidine Core Formation
The construction of the triazolopyrimidine core is the cornerstone of this synthetic route, relying on the cyclization of a pyrimidine precursor with a hydrazine or equivalent nitrogen source to form the fused ring system. The mechanism likely proceeds through an initial nucleophilic attack followed by dehydration to establish the aromatic character of the triazole ring, which is essential for the planar geometry required for intercalation or stacking interactions within the enzyme pocket. Subsequent functionalization at the C-5 position with a propylthio group enhances lipophilicity and may play a role in occupying a specific hydrophobic sub-pocket of the USP28 protein, thereby increasing binding affinity. The strategic placement of the amino group at the C-7 position, varied through different R groups, allows for hydrogen bonding interactions with key residues in the catalytic domain, which is supported by the observed high inhibition rates in the biological assays.
Impurity control is meticulously managed through the selection of reaction parameters, particularly during the diazotization step where temperature control between -10°C and 20°C is paramount to prevent over-reaction or decomposition of the sensitive diazo intermediate. The use of acetic acid as a solvent in this stage helps stabilize the reactive species, while the subsequent quenching and extraction protocols ensure the removal of inorganic salts and unreacted starting materials. This attention to detail in the process design results in intermediates that are sufficiently pure for the final coupling steps, reducing the burden on chromatographic purification and improving the overall mass balance of the synthesis. Such mechanistic understanding is vital for scaling up the process, as it highlights the critical process parameters that must be monitored to ensure batch-to-batch consistency and product quality.
How to Synthesize Triazolopyrimidine Derivatives Efficiently
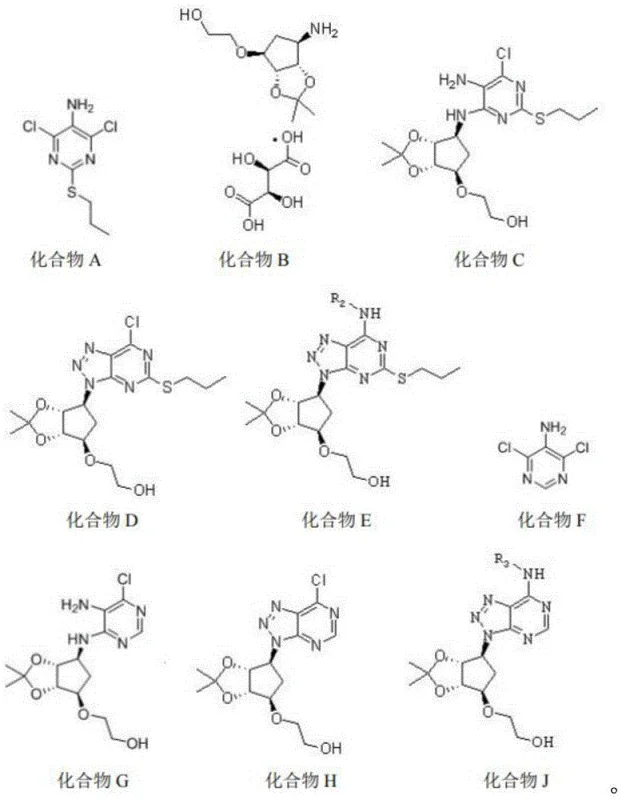
The synthesis of these high-purity pharmaceutical intermediates follows a logical sequence of transformations that can be adapted for both laboratory and pilot-scale production. The process begins with the formation of the chloro-substituted triazolopyrimidine core, which serves as a versatile electrophile for subsequent nucleophilic aromatic substitution. Detailed standardized synthesis steps are provided in the guide below, outlining the specific stoichiometry and workup procedures required to achieve optimal yields.
- Condense compound A or F with compound B in a alcoholic solvent at elevated temperatures (64-130°C) to form the triazolopyrimidine core intermediate.
- Perform diazotization on the intermediate using sodium nitrite in acetic acid at low temperatures (-10 to 20°C) to activate the ring for substitution.
- React the activated intermediate with specific amines (NH2-R) in acetonitrile to introduce the desired side chains, followed by sulfamoylation if required for Formula II or III.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the synthetic route described in this patent offers substantial benefits due to its reliance on commodity chemicals and straightforward unit operations. The starting materials, such as the pyrimidine precursors and various benzyl amines, are widely available in the global chemical market, reducing the risk of supply chain disruptions associated with exotic or proprietary reagents. This accessibility translates directly into enhanced supply chain reliability, as multiple qualified vendors can be sourced for raw materials, preventing single-source bottlenecks that often plague complex drug synthesis. Furthermore, the elimination of transition metal catalysts in the key coupling steps removes the need for expensive and technically demanding heavy metal scavenging processes, which significantly simplifies the purification workflow and lowers operational costs.
- Cost Reduction in Manufacturing: The process avoids the use of precious metal catalysts and operates at moderate temperatures, which drastically reduces energy consumption and equipment wear compared to high-pressure or cryogenic alternatives. By utilizing standard solvents like ethanol and acetonitrile, the recovery and recycling of these materials become economically feasible, contributing to substantial cost savings in large-scale production runs. The high yields reported for key intermediates, such as the 88% yield for Compound D in the examples, indicate a material-efficient process that minimizes waste generation and maximizes the output per batch.
- Enhanced Supply Chain Reliability: The modular nature of the synthesis allows for the decoupling of certain steps, meaning that key intermediates can be stockpiled or sourced independently to buffer against demand fluctuations. Since the reaction conditions are not overly sensitive to minor variations in moisture or oxygen (outside of specific inert atmosphere steps), the process is robust enough to be transferred between different manufacturing sites without extensive re-validation. This flexibility ensures continuous supply continuity, which is critical for maintaining clinical trial timelines and meeting regulatory commitments for drug substance availability.
- Scalability and Environmental Compliance: The absence of highly toxic reagents and the use of aqueous workups for salt removal align well with modern green chemistry principles and environmental regulations. The process generates waste streams that are easier to treat compared to those containing heavy metals or persistent organic pollutants, facilitating compliance with increasingly stringent environmental standards. Additionally, the scalability is supported by the use of standard reactor types, such as autoclaves for the initial heating steps and standard stirred tanks for the room temperature reactions, allowing for a seamless transition from kilogram to ton-scale production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these triazolopyrimidine derivatives. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation to ensure accuracy and relevance for industry stakeholders.
Q: What is the primary biological target of these triazolopyrimidine derivatives?
A: These compounds are designed as potent inhibitors of USP28 (Ubiquitin Specific Peptidase 28), a deubiquitinating enzyme implicated in tumor growth and cell cycle regulation.
Q: Which specific compounds demonstrated the highest inhibitory activity?
A: According to the patent data, Compound 29 and Compound 58 exhibited the optimal inhibitory capability, with IC50 values of approximately 1.15 μM and 1.20 μM respectively.
Q: Are the reaction conditions suitable for large-scale production?
A: Yes, the synthesis utilizes mild temperatures ranging from room temperature to 130°C and common industrial solvents like ethanol and acetonitrile, facilitating scalable manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Triazolopyrimidine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of USP28 inhibitors in the next generation of oncology therapeutics and are fully equipped to support your development needs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is smooth and efficient. We adhere to stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of triazolopyrimidine intermediates meets the highest quality standards required for pharmaceutical applications.
We invite you to contact our technical procurement team to discuss your specific requirements and to request a Customized Cost-Saving Analysis for your project. By partnering with us, you gain access to specific COA data and comprehensive route feasibility assessments that will help you optimize your supply chain and accelerate your drug development timeline. Let us be your trusted partner in bringing these innovative USP28 inhibitors from the laboratory to the patients who need them most.