Advanced Electrochemical Synthesis of Triazolopyrimidine Derivatives for Commercial Antitumor Drug Development
The pharmaceutical industry is constantly seeking novel scaffolds that can effectively disrupt microtubule dynamics to combat malignant tumors, which remain a leading cause of mortality globally. Patent CN113105459B introduces a significant advancement in this field by disclosing a series of 5-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-c]pyrimidine derivatives. These compounds are designed to function as potent tubulin inhibitors, specifically targeting the colchicine binding site, offering a promising alternative to existing therapies that often suffer from poor water solubility or severe toxicity. The core innovation lies not only in the biological efficacy of the molecules but also in the development of a greener, more efficient synthetic methodology that leverages electrochemical techniques. This approach addresses critical pain points in traditional organic synthesis, such as the reliance on hazardous oxidizing agents, thereby presenting a compelling value proposition for stakeholders focused on sustainable pharmaceutical intermediates manufacturing.
![General chemical structure of 5-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-c]pyrimidine derivatives showing the core scaffold and variable Ar group](/insights/img/triazolopyrimidine-synthesis-pharma-intermediate-supplier-20260306021522-01.png)
For R&D Directors evaluating new chemical entities, the structural versatility of this scaffold is paramount. The patent details a general formula where the Ar group can be varied extensively, including substituted phenyl rings and various indolyl moieties. This modularity allows for the fine-tuning of physicochemical properties, such as solubility and metabolic stability, which are often the downfall of potential drug candidates. By providing a robust platform for structure-activity relationship (SAR) studies, this technology enables the rapid identification of lead compounds with optimized therapeutic indices. The ability to synthesize these complex heterocycles efficiently is a key differentiator, as it lowers the barrier for entry into preclinical development pipelines, ensuring that promising antitumor agents can progress swiftly from the bench to biological evaluation without being bottlenecked by synthetic complexity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing fused triazolopyrimidine systems often rely heavily on stoichiometric amounts of chemical oxidants and reducing agents to drive redox transformations. These conventional methods frequently involve harsh reaction conditions, including extreme temperatures or the use of corrosive reagents, which can compromise the integrity of sensitive functional groups present in complex molecular architectures. Furthermore, the generation of substantial chemical waste associated with these redox agents poses significant environmental challenges and increases the cost of waste disposal, a critical factor in modern green chemistry mandates. From a process safety perspective, handling large quantities of strong oxidizers introduces inherent risks in a manufacturing setting, potentially leading to supply chain disruptions due to regulatory scrutiny or safety incidents. These limitations collectively hinder the scalability and economic viability of producing high-purity pharmaceutical intermediates on a commercial scale.
The Novel Approach
In stark contrast, the methodology outlined in the patent utilizes an electrochemical anodic oxidation strategy to achieve the necessary cyclization, effectively replacing chemical oxidants with electrons. This innovative approach operates under mild conditions, typically at room temperature or slightly elevated temperatures (25-40°C), using a constant voltage (e.g., 10V) in an undivided cell equipped with a graphite anode and platinum cathode. By avoiding the use of expensive and hazardous chemical oxidants, this method not only simplifies the operational workflow but also drastically reduces the environmental footprint of the synthesis. The subsequent step involves a Suzuki coupling reaction coupled with a Dimroth rearrangement, which seamlessly constructs the final triazolo[1,5-c]pyrimidine core. This tandem process exemplifies step economy, allowing for the rapid assembly of complex structures from readily available starting materials like 2-chloro-4-hydrazinopyrimidine and aryl aldehydes, thereby enhancing the overall efficiency of cost reduction in pharmaceutical intermediates manufacturing.
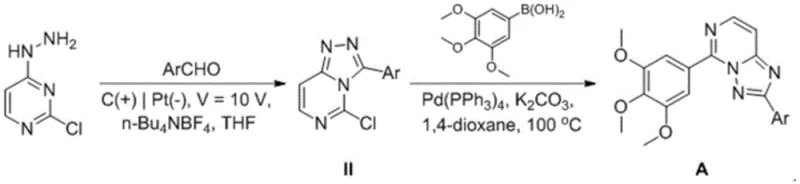
Mechanistic Insights into Electrochemical Cyclization and Suzuki-Dimroth Rearrangement
The mechanistic pathway begins with the condensation of 2-chloro-4-hydrazinopyrimidine and an aryl aldehyde to form a hydrazone intermediate in situ. Under electrochemical conditions, this intermediate undergoes anodic oxidation, facilitating an intramolecular cyclization to yield a 5-chloro-[1,2,4]triazolo[4,3-c]pyrimidine derivative. The use of tetra-n-butylammonium fluoroborate (n-Bu4NBF4) as a supporting electrolyte in tetrahydrofuran (THF) ensures efficient charge transfer while maintaining the stability of the reactive species. This electrochemical step is crucial as it avoids the over-oxidation or side reactions often seen with chemical oxidants, leading to cleaner reaction profiles and higher purity of the intermediate. The control of voltage, typically optimized between 5V and 20V, allows for precise modulation of the oxidation potential, ensuring selective transformation of the hydrazone without degrading the sensitive pyrimidine ring system.
Following the electrochemical step, the chloro-intermediate serves as an electrophile in a palladium-catalyzed Suzuki cross-coupling reaction with 3,4,5-trimethoxyphenylboronic acid. This step is particularly ingenious as it triggers a spontaneous Dimroth rearrangement, converting the initial [4,3-c] fused system into the thermodynamically more stable [1,5-c] isomer found in the final product. The reaction is conducted in 1,4-dioxane at 100°C using Pd(PPh3)4 as the catalyst and potassium carbonate as the base. This rearrangement is driven by the electronic properties of the substituents and the reaction conditions, effectively locking the molecule into the bioactive conformation required for tubulin binding. Understanding this dual mechanism is vital for commercial scale-up of complex pharmaceutical intermediates, as it highlights the robustness of the process and the minimal need for intermediate isolation, which streamlines the production timeline and reduces material loss.
How to Synthesize 5-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-c]pyrimidine Derivatives Efficiently
The synthesis protocol described offers a standardized pathway for producing these high-value antitumor agents. The process begins with the preparation of the electrochemical cell, where precise control of electrode surface area and electrolyte concentration is maintained to ensure reproducibility. Following the electrolysis, the crude intermediate is extracted and purified, typically via silica gel column chromatography, before being subjected to the Suzuki coupling conditions. The integration of these steps into a cohesive workflow minimizes handling time and exposure to air-sensitive reagents. For detailed operational parameters, including specific molar ratios, solvent volumes, and purification techniques, please refer to the standardized guide below which outlines the critical process controls necessary for achieving consistent quality.
- Perform electrochemical anodic oxidation of 2-chloro-4-hydrazinopyrimidine and aryl aldehyde using a carbon anode and platinum cathode in THF with n-Bu4NBF4 electrolyte at 10V.
- Conduct Suzuki coupling of the resulting chloro-intermediate with 3,4,5-trimethoxyphenylboronic acid using Pd(PPh3)4 catalyst and K2CO3 base in 1,4-dioxane at 100°C.
- Purify the final triazolopyrimidine derivative via column chromatography after aqueous workup and solvent removal.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this electrochemical synthesis route presents tangible strategic benefits that extend beyond mere technical novelty. The elimination of stoichiometric chemical oxidants translates directly into a simplified supply chain, as there is no longer a dependency on sourcing, storing, and disposing of hazardous oxidizing agents. This reduction in raw material complexity inherently lowers the risk of supply disruptions caused by regulatory changes or vendor shortages associated with specialized reagents. Furthermore, the mild reaction conditions reduce the energy consumption required for heating or cooling, contributing to a lower overall carbon footprint and aligning with corporate sustainability goals. These factors collectively enhance the resilience of the supply chain, ensuring a more reliable flow of critical pharmaceutical intermediates to downstream drug manufacturers.
- Cost Reduction in Manufacturing: The electrochemical method fundamentally alters the cost structure of the synthesis by replacing expensive chemical reagents with electricity, which is generally a cheaper and more stable utility cost. By avoiding the purchase of high-cost oxidants and the associated waste treatment fees, the overall cost of goods sold (COGS) is significantly optimized. Additionally, the high atom economy of the Suzuki-Dimroth sequence minimizes raw material waste, further driving down production costs. This economic efficiency makes the technology highly attractive for large-scale manufacturing, where even marginal savings per kilogram can result in substantial financial benefits over the lifecycle of a drug product.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, such as 2-chloro-4-hydrazinopyrimidine and various aryl aldehydes, are commodity chemicals that are widely available from multiple global suppliers. This abundance mitigates the risk of single-source dependency, a common vulnerability in pharmaceutical supply chains. The robustness of the electrochemical process also means that production is less susceptible to variations in reagent quality, ensuring consistent output even when sourcing from different vendors. This reliability is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines required by clinical and commercial partners.
- Scalability and Environmental Compliance: Scaling electrochemical reactions is increasingly feasible with modern flow chemistry technologies, allowing for a seamless transition from laboratory gram-scale to multi-ton commercial production. The green nature of this process, characterized by reduced hazardous waste and safer operating conditions, simplifies compliance with stringent environmental regulations. This ease of compliance accelerates the regulatory approval process for manufacturing sites and reduces the administrative burden on EHS teams. Consequently, companies can bring products to market faster while maintaining a strong commitment to environmental stewardship and operational safety.
Frequently Asked Questions (FAQ)
The following questions address common inquiries regarding the technical feasibility and commercial potential of this synthesis technology. They are derived from the specific experimental data and beneficial effects reported in the patent documentation, providing clarity on how this method compares to industry standards. Understanding these details is essential for making informed decisions about integrating this technology into existing production portfolios or R&D pipelines.
Q: What are the key advantages of the electrochemical method described in CN113105459B?
A: The electrochemical method eliminates the need for stoichiometric chemical oxidants and reducing agents, significantly reducing chemical waste and cost while simplifying the operation steps for complex molecule construction.
Q: What is the biological activity profile of these triazolopyrimidine derivatives?
A: These compounds act as tubulin inhibitors targeting the colchicine binding site, demonstrating significant inhibition against HeLa, MCF-7, and HCT116 tumor cell lines with low toxicity to normal cells.
Q: Is this synthesis route scalable for commercial production?
A: Yes, the use of mild reaction conditions, readily available starting materials, and the avoidance of expensive catalysts in the first step makes this route highly suitable for commercial scale-up and supply chain reliability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Triazolopyrimidine Derivatives Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis and contract development, possessing the technical expertise to translate complex patent methodologies like CN113105459B into robust commercial processes. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and efficiency. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of high-purity pharmaceutical intermediates meets the highest international standards. Our commitment to quality and consistency makes us an ideal partner for navigating the challenges of bringing novel antitumor agents from concept to reality.
We invite you to engage with our technical procurement team to discuss how we can support your specific requirements. Whether you need a Customized Cost-Saving Analysis for your current supply chain or require specific COA data and route feasibility assessments for these triazolopyrimidine derivatives, we are ready to provide comprehensive solutions. Contact us today to explore how our advanced manufacturing capabilities can accelerate your drug development timeline and secure your supply of critical therapeutic intermediates.