Advanced Enantioselective Synthesis of Quinoline Derivatives for Commercial API Production
Advanced Enantioselective Synthesis of Quinoline Derivatives for Commercial API Production
The pharmaceutical industry's demand for high-purity respiratory therapeutics has driven significant innovation in the synthesis of beta-adrenoceptor agonists. Patent CN1968927B introduces a groundbreaking enantioselective method for preparing 8-substituted oxy-5-((R)-2-halo-1-hydroxy-ethyl)-(1H)-quinolin-2-one derivatives. These compounds serve as critical intermediates for potent bronchodilators used in treating asthma and chronic obstructive pulmonary disease (COPD). Unlike traditional racemic syntheses that require costly and yield-reducing resolution steps, this patented methodology leverages advanced chiral transition metal catalysis to directly access the biologically active (R)-enantiomer. For R&D directors and procurement specialists, this represents a paradigm shift towards more efficient, sustainable, and economically viable manufacturing processes for complex heterocyclic APIs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinoline-based beta-agonists relied heavily on non-stereoselective pathways. Conventional routes often involved the reduction of alpha-halo ketones using achiral reducing agents like sodium borohydride, resulting in a racemic mixture of (R) and (S) enantiomers. Since biological activity is typically confined to a single enantiomer, manufacturers were forced to employ chiral resolution techniques, such as diastereomeric salt formation or chiral chromatography, to isolate the active component. These downstream purification steps are inherently inefficient, theoretically capping the maximum yield at 50% and generating substantial chemical waste. Furthermore, the separation of closely related stereoisomers often requires multiple recrystallizations, driving up solvent consumption, extending production lead times, and complicating the supply chain for high-purity pharmaceutical intermediates.
The Novel Approach
The methodology disclosed in CN1968927B circumvents these inefficiencies by integrating asymmetric transfer hydrogenation directly into the synthetic sequence. By reacting a 5-(alpha-haloacetyl)-8-substituted oxy-(1H)-quinolin-2-one with a reducing agent in the presence of a specific chiral Ruthenium catalyst and a base, the process selectively generates the 8-substituted oxy-5-((R)-2-halo-1-hydroxy-ethyl) derivative. This direct enantioselective transformation eliminates the formation of the unwanted (S)-isomer at the source, thereby removing the need for subsequent resolution. The process flow is streamlined, converting a multi-step resolution-heavy workflow into a concise catalytic cycle. This not only enhances the theoretical yield but also simplifies the impurity profile, making downstream processing significantly more robust and predictable for commercial scale-up.
Mechanistic Insights into Ru-Catalyzed Asymmetric Transfer Hydrogenation
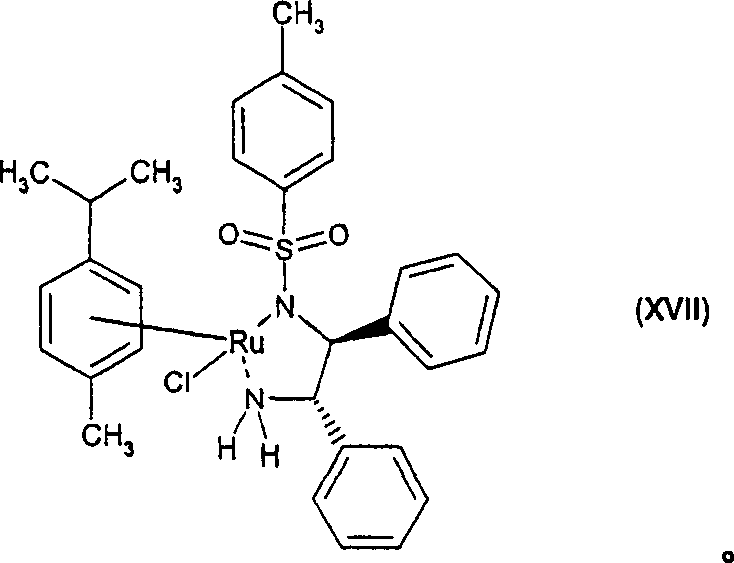
The core of this technological breakthrough lies in the utilization of Noyori-type chiral Ruthenium complexes. The patent specifies catalysts having Formula I or II, where M is preferably Ruthenium, coordinated with chiral diamine ligands such as (1S,2S)-N-p-toluenesulfonyl-1,2-diphenylethylenediamine (TsDPEN). In the presence of a hydrogen donor like formic acid and a tertiary amine base, these precatalysts generate an active Ruthenium hydride species in situ. This active species facilitates the transfer of a hydride ion and a proton to the prochiral ketone substrate in a concerted manner. The chiral environment created by the diamine ligand dictates the facial selectivity of the hydride attack, ensuring the exclusive formation of the (R)-alcohol configuration. This mechanism operates under mild conditions, typically between 0°C and 50°C, avoiding the harsh temperatures that can degrade sensitive quinoline scaffolds.
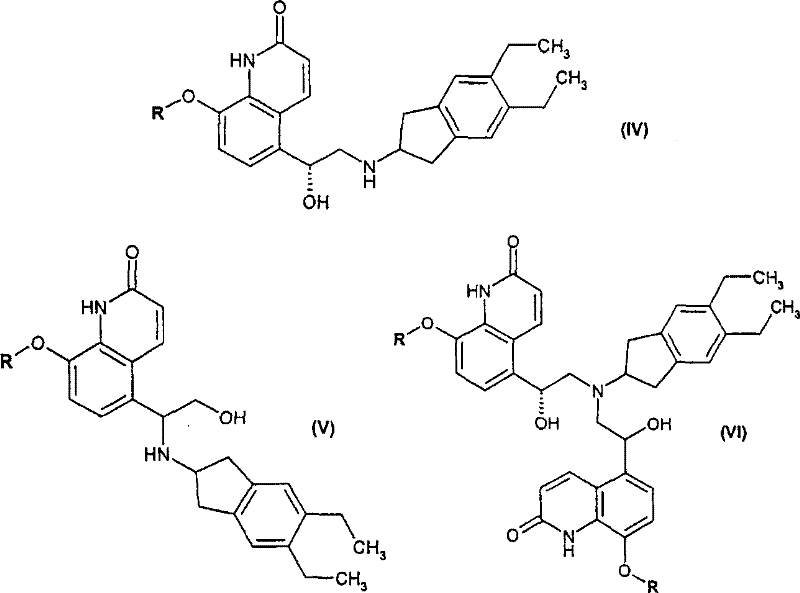
Beyond stereocontrol, the process demonstrates exceptional impurity management capabilities. The reaction conditions are tuned to minimize side reactions such as dehalogenation or over-reduction. Following the asymmetric reduction, the intermediate is cyclized to an epoxide using a base like potassium tert-butoxide. This epoxide serves as a versatile electrophile for the subsequent ring-opening reaction with 2-amino-5,6-diethylindane. The patent highlights that controlling the stoichiometry and temperature during this coupling step is crucial to favoring the formation of the desired amino-alcohol (Formula IV) over regioisomeric byproducts (Formula V and VI). The ability to steer the reaction towards the target molecule through precise parameter control underscores the robustness of this synthetic design for industrial applications.
How to Synthesize 8-Substituted Oxy-5-((R)-2-halo-1-hydroxy-ethyl)-(1H)-quinolin-2-one Efficiently
The synthesis begins with the Friedel-Crafts acylation of 8-hydroxy-(1H)-quinolin-2-one to install the acetyl group at the 5-position, followed by protection of the phenolic oxygen and halogenation of the side chain. The pivotal step involves the enantioselective reduction using the chiral Ruthenium catalyst system described above. Once the chiral alcohol is obtained, it is converted to an epoxide and coupled with the indane amine fragment. The final product is isolated as a stable salt, such as the benzoate or maleate, through controlled crystallization.
- Perform Friedel-Crafts acylation of 8-hydroxy-(1H)-quinolin-2-one using acetic anhydride and Lewis acid to generate 5-acetyl-8-hydroxy-(1H)-quinolin-2-one.
- Protect the 8-hydroxy group and halogenate the acetyl side chain to form 5-(alpha-haloacetyl)-8-substituted oxy-(1H)-quinolin-2-one.
- Execute enantioselective reduction using a chiral Ruthenium catalyst and formic acid/triethylamine to yield the (R)-halo-hydroxy-ethyl intermediate.
- Cyclize the halo-hydroxy intermediate into an epoxide using base, then react with 2-amino-5,6-diethylindane to form the final salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this enantioselective technology offers profound strategic benefits beyond mere chemical elegance. The elimination of chiral resolution steps translates directly into a drastic reduction in raw material consumption and waste disposal costs. By avoiding the loss of 50% of the material inherent in racemic resolutions, the effective throughput of the manufacturing plant is nearly doubled without increasing reactor volume. This efficiency gain allows for significant cost reduction in pharmaceutical intermediate manufacturing, enabling more competitive pricing for downstream API producers. Furthermore, the simplified process flow reduces the number of unit operations, lowering energy consumption and labor requirements associated with batch processing and quality control testing.
- Cost Reduction in Manufacturing: The catalytic nature of the chiral induction means that expensive chiral auxiliaries are not consumed stoichiometrically but are recycled within the catalytic cycle. The use of formic acid as a hydrogen source is economically superior to high-pressure hydrogenation, as it removes the need for specialized high-pressure reactor infrastructure and the safety costs associated with handling bulk hydrogen gas. This shift to ambient pressure transfer hydrogenation lowers capital expenditure barriers and operational risks, contributing to substantial cost savings in the overall production budget.
- Enhanced Supply Chain Reliability: The reagents employed in this process, including the Ruthenium precursors, formic acid, and common organic solvents like acetone and ethyl acetate, are commercially available from multiple global suppliers. This diversification of the supply base mitigates the risk of single-source bottlenecks that often plague specialized chiral reagent markets. Additionally, the robustness of the reaction conditions ensures consistent batch-to-batch reproducibility, which is critical for maintaining uninterrupted supply lines to API manufacturers who operate on tight just-in-time schedules.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing solvents and conditions that are easily managed in large-scale stainless steel reactors. The avoidance of cryogenic temperatures and hazardous reagents simplifies the engineering controls required for safe operation. From an environmental perspective, the atom economy of the asymmetric reduction is superior to classical resolution methods, resulting in a lower E-factor (mass of waste per mass of product). This aligns with modern green chemistry principles and helps pharmaceutical companies meet increasingly stringent regulatory requirements regarding solvent emissions and chemical waste disposal.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. Understanding these details is essential for evaluating the feasibility of integrating this technology into existing production portfolios.
Q: What is the primary advantage of the chiral Ruthenium catalyst described in CN1968927B?
A: The chiral Ruthenium catalyst, specifically complexes like RuCl[(1S,2S)-p-TsN-CH(C6H5)CH(C6H5)-NH2](η6-p-cymene), enables highly enantioselective transfer hydrogenation. This eliminates the need for difficult chiral resolution steps required in racemic synthesis, significantly improving overall yield and optical purity of the final bronchodilator intermediate.
Q: How does this process address impurity control in quinoline synthesis?
A: The patent outlines specific crystallization and salt formation protocols, particularly using benzoic acid or maleic acid, to selectively isolate the desired isomer (Formula IV) while minimizing byproducts like Formula V and VI. The use of specific solvents like ethanol and controlled temperature profiles during salt formation ensures high area percent purity exceeding 96%.
Q: Is this synthetic route scalable for industrial API manufacturing?
A: Yes, the process utilizes robust reagents such as formic acid, triethylamine, and common organic solvents like acetone and ethyl acetate. The avoidance of cryogenic conditions and the use of stable transition metal catalysts make the route highly amenable to scale-up from kilogram to multi-ton production levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinoline Derivatives Supplier
The synthesis of complex heterocyclic intermediates like those described in CN1968927B requires a partner with deep technical expertise and proven manufacturing capabilities. NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of quinoline derivative meets the exacting standards required for respiratory drug development. We understand the critical nature of chirality in pharmaceutical efficacy and have optimized our processes to deliver high enantiomeric excess consistently.
We invite potential partners to engage with our technical procurement team to discuss how this advanced enantioselective route can be tailored to your specific project needs. By leveraging our expertise, you can secure a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this catalytic method. Contact us today to request specific COA data and route feasibility assessments, and let us help you accelerate your path to market with reliable, high-quality pharmaceutical intermediates.