Advanced Manufacturing of Chiral Imidazolidinone Derivatives for Neurological Therapeutics
Introduction to Patent CN1135216A and Neurological Applications
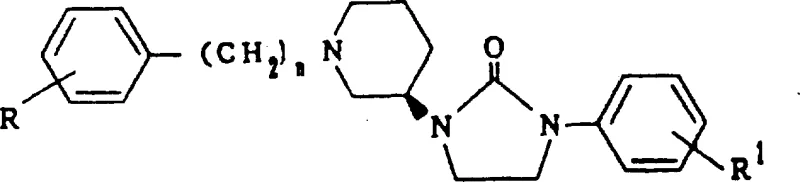
The pharmaceutical landscape for treating senile dementia and Alzheimer's disease has been significantly advanced by the discoveries detailed in patent CN1135216A, which discloses novel optically active imidazolidinone derivatives. These compounds exhibit potent cholinergic activity, specifically targeting muscarinic M1 receptors, which are critically linked to memory formation and cognitive function. Unlike previous generations of acetylcholinesterase inhibitors that suffered from narrow therapeutic windows and severe side effects, these new derivatives offer a targeted approach to activating central cholinergic pathways. The core innovation lies in the stereochemical configuration, where the specific R-enantiomer demonstrates vastly superior pharmacological profiles compared to its S-counterpart or racemic mixtures. This distinction is not merely academic; it represents a fundamental shift in how we approach the synthesis of CNS-active agents, prioritizing enantiomeric purity to maximize efficacy while minimizing neurotoxicity. As a reliable pharmaceutical intermediate supplier, understanding the nuances of this patent is essential for developing next-generation therapeutics that address the growing global burden of neurodegenerative disorders.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of imidazolidinone scaffolds often relied on non-stereoselective pathways that yielded racemic mixtures, as seen in earlier literature such as US Patent 3,459,757. These conventional methods failed to distinguish between the biological activities of individual enantiomers, leading to drug candidates with compromised safety profiles. The presence of the inactive or toxic S-isomer in racemic formulations necessitates higher dosing to achieve therapeutic levels of the active component, thereby increasing the risk of adverse events such as convulsions and general CNS depression. Furthermore, the downstream processing required to separate enantiomers from a racemic bulk is notoriously inefficient, involving complex chromatography or repeated crystallization steps that drastically reduce overall yield and inflate manufacturing costs. From a supply chain perspective, relying on racemic synthesis introduces significant variability in batch quality and complicates regulatory approval processes, as agencies increasingly demand rigorous characterization of all stereoisomers present in a drug substance.
The Novel Approach
The methodology outlined in CN1135216A circumvents these issues by employing a chiral pool strategy, initiating the synthesis with optically pure starting materials such as resolved (R)-3-piperidinecarboxylic acid ethyl ester. This approach ensures that the stereochemistry is established at the very beginning of the synthetic route, propagating through subsequent transformations like the Hofmann rearrangement and reductive amination without loss of optical integrity. By focusing on the exclusive production of the R-isomer, manufacturers can achieve a product with approximately 120 times the M1 receptor affinity of the S-isomer, effectively eliminating the toxic load associated with the unwanted enantiomer. This strategic pivot not only enhances the therapeutic index but also streamlines the purification process, as the need for chiral resolution of the final product is obviated. For procurement teams, this translates to a more robust and predictable manufacturing process capable of delivering high-purity active pharmaceutical ingredients (APIs) with consistent biological performance.
Mechanistic Insights into Carbonyl Insertion and Cyclization
The core chemical transformation in this patent involves the construction of the imidazolidinone ring via a carbonyl insertion reaction, a critical step that defines the structural integrity of the final molecule. This cyclization is typically achieved by reacting an N-substituted ethylenediamine precursor with carbonylating agents such as N,N'-carbonyldiimidazole (CDI), phosgene, or diethyl carbonate. The reaction mechanism proceeds through the nucleophilic attack of the secondary amine on the electrophilic carbonyl carbon of the activating agent, forming a reactive intermediate that subsequently undergoes intramolecular cyclization with the adjacent urea or amide nitrogen. The choice of solvent and temperature is paramount; reactions are conducted in solvents like tetrahydrofuran or dioxane at temperatures ranging from 0°C to 150°C, depending on the specific reactivity of the substituents. This flexibility allows for the optimization of reaction kinetics to favor the formation of the five-membered imidazolidinone ring while suppressing potential polymerization or degradation pathways. The precision required in this step underscores the importance of process control in maintaining the high purity standards demanded for neurological drugs.
Another pivotal mechanistic feature is the preservation of chirality during the formation of the 3-amino-1-aralkylpiperidine scaffold. The synthesis leverages the Hofmann rearrangement of optically active amides, a reaction known for its ability to migrate the amino group with retention of configuration at the chiral center. This is followed by N-alkylation with aralkyl halides, a step that must be carefully managed to prevent racemization via enolization or SN1 pathways. The use of mild bases and controlled temperatures during alkylation ensures that the stereochemical information encoded in the starting piperidine derivative is faithfully transmitted to the final imidazolidinone product. Understanding these mechanistic subtleties is vital for R&D directors aiming to scale these reactions, as even minor deviations in pH or thermal history can lead to erosion of enantiomeric excess, compromising the drug's safety and efficacy profile.
How to Synthesize Optically Active Imidazolidinone Derivatives Efficiently
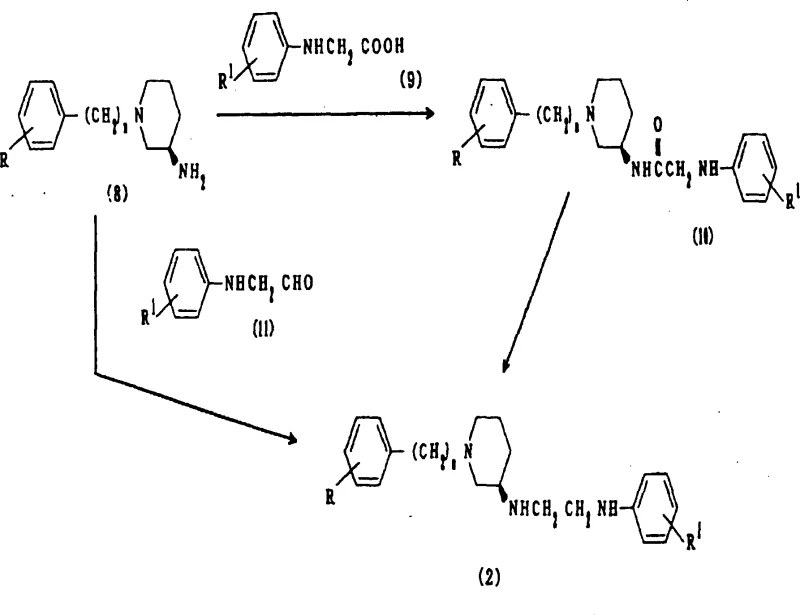
The efficient production of these high-value intermediates requires a meticulously planned sequence of reactions that balances yield, purity, and operational simplicity. The process begins with the preparation of the chiral amine backbone, followed by coupling with phenylglycine derivatives and final ring closure. Each step has been optimized in the patent examples to demonstrate feasibility on a laboratory scale, providing a solid foundation for industrial adaptation. The following guide outlines the standardized synthetic steps derived from the patent data, offering a clear roadmap for technical teams looking to implement this chemistry.
- Preparation of the chiral diamine precursor through reductive amination of optically active 3-amino-1-aralkylpiperidine with N-phenylglycine derivatives.
- Cyclization of the diamine intermediate using carbonylating agents such as N,N'-carbonyldiimidazole (CDI) or phosgene equivalents at elevated temperatures.
- Purification of the final optically active imidazolidinone derivative via column chromatography and recrystallization to ensure high enantiomeric excess.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting the synthesis routes described in CN1135216A offers substantial strategic advantages for pharmaceutical manufacturers, particularly in terms of cost structure and supply chain resilience. By eliminating the need for post-synthesis chiral resolution, the process significantly reduces the consumption of expensive resolving agents and solvents, leading to a leaner and more cost-effective manufacturing workflow. The use of readily available chiral building blocks, such as resolved pipecolic acid derivatives, ensures a stable supply of raw materials, mitigating the risks associated with sourcing exotic or proprietary catalysts. Furthermore, the robustness of the carbonyl insertion step allows for scalable production without the need for specialized high-pressure equipment, facilitating easier technology transfer from pilot plants to commercial-scale facilities. These factors collectively contribute to a lower cost of goods sold (COGS) and a more competitive market position for the final therapeutic agent.
- Cost Reduction in Manufacturing: The streamlined synthetic route avoids the inefficiencies inherent in racemic synthesis, where up to 50% of the material (the S-isomer) is essentially waste. By synthesizing only the active R-isomer from the outset, raw material utilization is maximized, and the burden on waste management systems is drastically reduced. Additionally, the avoidance of heavy metal catalysts in favor of organic reagents like CDI simplifies the purification process, removing the need for costly metal scavenging steps and reducing the environmental footprint of the manufacturing site.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals and standard organic transformations ensures that the supply chain is less vulnerable to disruptions caused by the scarcity of specialized reagents. The synthetic pathway is modular, allowing for the interchangeability of certain substituents without altering the core process flow, which provides flexibility in sourcing precursors. This adaptability is crucial for maintaining continuous production schedules and meeting the fluctuating demands of the global pharmaceutical market, ensuring that critical medications for Alzheimer's treatment remain available to patients.
- Scalability and Environmental Compliance: The reaction conditions described, such as atmospheric pressure cyclization and moderate temperature ranges, are inherently safer and easier to scale than high-energy processes. This reduces the capital expenditure required for reactor infrastructure and lowers the operational risks associated with thermal runaways. Moreover, the generation of benign byproducts like imidazole or carbon dioxide aligns with modern green chemistry principles, facilitating regulatory compliance and reducing the costs associated with hazardous waste disposal and environmental remediation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these chiral imidazolidinone derivatives. The answers are derived directly from the experimental data and claims within the patent documentation, providing accurate insights for stakeholders evaluating this technology. Understanding these details is key to making informed decisions about process adoption and partnership opportunities.
Q: Why is the optical purity critical for imidazolidinone derivatives in dementia treatment?
A: The patent data reveals that the R-isomer exhibits approximately 120 times higher M1 muscarinic activity compared to the S-isomer. Furthermore, the S-isomer and racemic mixtures demonstrate significant toxicity, including convulsive symptoms at high doses, whereas the pure R-isomer shows a superior safety profile even at 1200 mg/kg.
Q: What are the key challenges in scaling up the synthesis of these chiral intermediates?
A: Maintaining stereochemical integrity during the Hofmann rearrangement and subsequent alkylation steps is crucial. The process requires precise control of reaction temperatures (0-25°C for coupling, up to 150°C for cyclization) and the use of specific resolving agents like dibenzoyl tartaric acid to ensure high enantiomeric purity.
Q: How does this novel synthesis route improve upon conventional racemic methods?
A: Conventional methods often produce racemic mixtures requiring difficult and costly separation. This patented approach utilizes optically active starting materials (like resolved ethyl pipecolate) early in the synthesis, ensuring the final product is inherently chiral and eliminating the need for downstream resolution of the toxic S-enantiomer.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Imidazolidinone Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the optically active imidazolidinone derivatives described in CN1135216A for the treatment of neurodegenerative diseases. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory discovery to market-ready therapy is seamless and efficient. We are committed to delivering high-purity intermediates that meet stringent purity specifications, utilizing our rigorous QC labs to verify enantiomeric excess and impurity profiles at every stage of production. Our state-of-the-art facilities are equipped to handle the specific thermal and solvent requirements of the carbonyl insertion and Hofmann rearrangement steps, guaranteeing consistent batch-to-batch quality.
We invite you to collaborate with us to optimize your supply chain and accelerate your drug development timeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can support your mission to bring life-changing treatments for Alzheimer's disease to patients worldwide.