Advanced Palladium-Catalyzed Carbonylation for Scalable N-Acyl Indole Manufacturing
Advanced Palladium-Catalyzed Carbonylation for Scalable N-Acyl Indole Manufacturing
The structural motif of N-acyl indoles represents a cornerstone in modern medicinal chemistry, serving as the critical pharmacophore in a vast array of bioactive molecules ranging from anti-inflammatory agents like Indomethacin to potent anti-HIV drugs such as Delavirdine. As depicted in the structural diversity of known pharmaceuticals, the ability to efficiently construct this scaffold is paramount for drug discovery and process development. 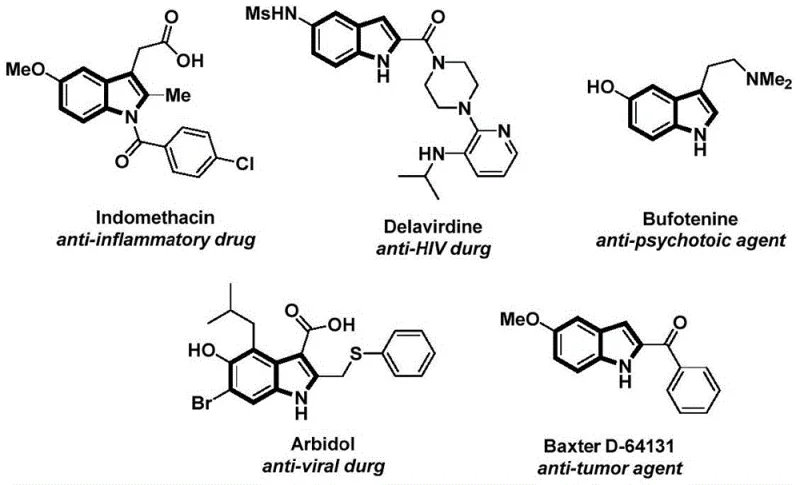 Recent advancements disclosed in Chinese Patent CN112898192B introduce a transformative methodology for the preparation of these compounds, utilizing a palladium-catalyzed carbonylation cyclization strategy. This innovation addresses long-standing challenges in synthetic efficiency and operational safety, offering a robust pathway for the production of high-purity pharmaceutical intermediates. By leveraging a solid carbon monoxide surrogate, this technology bypasses the logistical nightmares associated with gaseous CO, positioning it as a highly attractive solution for reliable pharmaceutical intermediate suppliers seeking to optimize their manufacturing portfolios.
Recent advancements disclosed in Chinese Patent CN112898192B introduce a transformative methodology for the preparation of these compounds, utilizing a palladium-catalyzed carbonylation cyclization strategy. This innovation addresses long-standing challenges in synthetic efficiency and operational safety, offering a robust pathway for the production of high-purity pharmaceutical intermediates. By leveraging a solid carbon monoxide surrogate, this technology bypasses the logistical nightmares associated with gaseous CO, positioning it as a highly attractive solution for reliable pharmaceutical intermediate suppliers seeking to optimize their manufacturing portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of N-acyl indoles has often relied on methodologies that impose significant burdens on process safety and scalability. Classical approaches frequently necessitate the use of carbon monoxide gas, which is not only highly toxic but also requires specialized high-pressure reactors and rigorous safety protocols to prevent leakage and exposure. Furthermore, many conventional routes involve multi-step sequences where the indole core is constructed first, followed by a separate acylation step, leading to cumulative yield losses and increased waste generation. These legacy methods often suffer from poor atom economy and limited functional group tolerance, forcing chemists to employ protecting group strategies that further elongate the synthetic timeline. For procurement managers and supply chain heads, these inefficiencies translate directly into higher costs of goods sold (COGS) and extended lead times, creating bottlenecks in the supply of critical active pharmaceutical ingredients (APIs).
The Novel Approach
In stark contrast, the methodology outlined in Patent CN112898192B presents a streamlined, one-pot solution that elegantly merges carbonylation and cyclization into a single operational sequence. 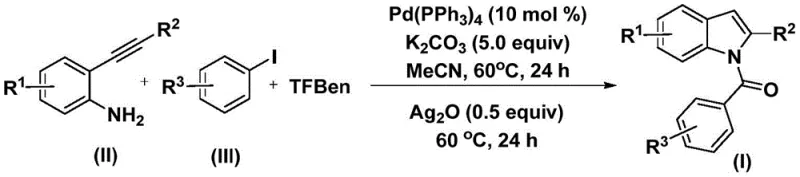 This novel approach utilizes 2-alkynylanilines and aryl iodides as readily available starting materials, reacting them in the presence of a palladium catalyst and a solid CO source known as TFBen (1,3,5-tricarboxylic acid phenol ester). The reaction proceeds under remarkably mild conditions, specifically at 60°C in acetonitrile, avoiding the extreme temperatures and pressures typical of traditional carbonylations. The introduction of silver oxide in the second stage facilitates an oxidative cyclization that rapidly closes the indole ring with high efficiency. This telescoped process not only simplifies the workflow but also drastically reduces the environmental footprint by minimizing solvent usage and purification steps, representing a significant leap forward in cost reduction in API manufacturing.
This novel approach utilizes 2-alkynylanilines and aryl iodides as readily available starting materials, reacting them in the presence of a palladium catalyst and a solid CO source known as TFBen (1,3,5-tricarboxylic acid phenol ester). The reaction proceeds under remarkably mild conditions, specifically at 60°C in acetonitrile, avoiding the extreme temperatures and pressures typical of traditional carbonylations. The introduction of silver oxide in the second stage facilitates an oxidative cyclization that rapidly closes the indole ring with high efficiency. This telescoped process not only simplifies the workflow but also drastically reduces the environmental footprint by minimizing solvent usage and purification steps, representing a significant leap forward in cost reduction in API manufacturing.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cyclization
The mechanistic elegance of this transformation lies in its sequential catalytic cycle, which ensures high selectivity and minimizes side reactions. The process initiates with the oxidative addition of the palladium(0) catalyst into the carbon-iodine bond of the aryl iodide, generating a reactive aryl-palladium(II) intermediate. Subsequently, the carbon monoxide released in situ from the thermal decomposition of TFBen inserts into the palladium-carbon bond, forming an acyl-palladium species. This key intermediate then undergoes nucleophilic attack by the amine group of the 2-alkynylaniline, followed by reductive elimination to yield an amide precursor. The final and crucial step involves the addition of silver oxide, which acts as an oxidant to promote the intramolecular cyclization of the alkyne moiety, ultimately delivering the N-acyl indole product while regenerating the active palladium species. This detailed understanding of the catalytic cycle allows R&D directors to fine-tune reaction parameters for optimal impurity control.
From an impurity profiling perspective, the use of a solid CO surrogate like TFBen provides a controlled, steady release of carbon monoxide, preventing the local concentration spikes that can lead to homocoupling side products or over-carbonylation. The mild reaction temperature of 60°C further suppresses thermal degradation pathways that are common in harsher acidic or basic cyclization methods. Moreover, the broad substrate compatibility demonstrated in the patent data indicates that electron-donating and electron-withdrawing groups on the aromatic rings do not significantly hinder the catalytic turnover. This robustness ensures that the final crude product contains a cleaner profile of by-products, simplifying downstream purification and ensuring that the resulting high-purity pharmaceutical intermediates meet stringent regulatory specifications for residual metals and organic impurities.
How to Synthesize N-Acyl Indole Efficiently
The practical implementation of this synthesis is designed for ease of execution in standard laboratory and pilot plant settings. The protocol begins by charging a reaction vessel with the palladium catalyst, specifically tetrakis(triphenylphosphine)palladium, along with potassium carbonate as the base and TFBen as the carbonyl source. To this mixture, the 2-alkynylaniline and aryl iodide substrates are added in acetonitrile solvent. The detailed standardized synthesis steps for this efficient route are provided in the guide below.
- Combine palladium catalyst (Pd(PPh3)4), potassium carbonate, carbon monoxide substitute (TFBen), 2-alkynylaniline, and aryl iodide in an organic solvent such as acetonitrile.
- Heat the reaction mixture at 60°C for 24 hours to facilitate the initial carbonylation and coupling steps.
- Add silver oxide (Ag2O) to the mixture and continue heating at 60°C for an additional 24 hours to promote oxidative cyclization, followed by filtration and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, this patented process offers compelling strategic advantages that go beyond mere chemical novelty. The shift from gaseous CO to a solid surrogate fundamentally alters the risk profile of the manufacturing process, removing the need for expensive high-pressure autoclaves and specialized gas handling infrastructure. This reduction in capital expenditure (CAPEX) allows for more flexible production scheduling and the potential for decentralized manufacturing closer to key markets. Furthermore, the reliance on commercially available starting materials such as aryl iodides and substituted anilines ensures a stable supply chain, mitigating the risks associated with sourcing exotic or custom-synthesized reagents that often plague complex API campaigns.
- Cost Reduction in Manufacturing: The elimination of high-pressure gas equipment and the consolidation of multiple synthetic steps into a single pot significantly lowers operational expenditures. By avoiding the need for specialized safety containment for toxic gases, facilities can reduce insurance premiums and compliance costs. Additionally, the high reaction efficiency and yields reported (ranging up to 82% in optimized examples) mean less raw material is wasted per kilogram of product, directly improving the overall material cost structure. The simplified work-up procedure, involving simple filtration and chromatography, further reduces labor hours and solvent consumption, contributing to substantial cost savings in pharmaceutical intermediate manufacturing.
- Enhanced Supply Chain Reliability: The use of robust, shelf-stable reagents like TFBen and common palladium catalysts ensures that production is not held hostage by the logistics of hazardous gas delivery. This stability allows for better inventory management and just-in-time manufacturing capabilities. The broad functional group tolerance means that a single platform technology can be adapted to produce a wide library of N-acyl indole derivatives without needing to requalify entirely new processes for each analog. This flexibility is crucial for reducing lead time for high-purity pharmaceutical intermediates, enabling faster response to market demands and clinical trial material requirements.
- Scalability and Environmental Compliance: Operating at a mild temperature of 60°C significantly reduces energy consumption compared to high-temperature reflux conditions, aligning with green chemistry principles and corporate sustainability goals. The process generates fewer hazardous by-products, simplifying waste treatment and disposal protocols. The scalability of the reaction is supported by the homogeneous nature of the catalytic system, which translates well from gram-scale optimization to multi-kilogram production batches. This ease of scale-up ensures that the commercial scale-up of complex pharmaceutical intermediates can be achieved with minimal technical risk, guaranteeing a continuous supply for downstream formulation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method, derived directly from the experimental data and specifications within the patent documentation. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this route for their specific pipeline candidates. The answers reflect the proven capabilities of the technology as demonstrated in the provided examples.
Q: What is the primary advantage of using TFBen in this synthesis?
A: TFBen (1,3,5-tricarboxylic acid phenol ester) serves as a solid carbon monoxide surrogate, eliminating the safety hazards and specialized equipment required for handling toxic high-pressure CO gas, thereby simplifying the operational protocol.
Q: What are the typical reaction conditions for this N-acyl indole preparation?
A: The reaction typically proceeds in acetonitrile at a mild temperature of 60°C. It involves a two-stage heating process: 24 hours for the initial coupling and carbonylation, followed by another 24 hours after the addition of silver oxide for cyclization.
Q: Does this method support diverse functional groups on the substrate?
A: Yes, the method demonstrates excellent substrate compatibility, tolerating various substituents such as methyl, methoxy, halogens (F, Cl, Br), and tert-butyl groups on both the aniline and aryl iodide components without significant yield loss.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Acyl Indole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and safe synthetic routes in the development of next-generation therapeutics. Our team of expert chemists has extensively evaluated the Pd-catalyzed carbonylation technology described in CN112898192B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are committed to delivering N-acyl indole intermediates that adhere to stringent purity specifications, utilizing our rigorous QC labs to ensure every batch meets the highest standards of quality and consistency required by global regulatory bodies.
We invite you to collaborate with us to leverage this advanced chemistry for your upcoming projects. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your drug development timeline while optimizing your overall budget.