Advanced Rhodium Catalysis for Commercial Scale-up of Complex Chiral Isoindolinones
Advanced Rhodium Catalysis for Commercial Scale-up of Complex Chiral Isoindolinones
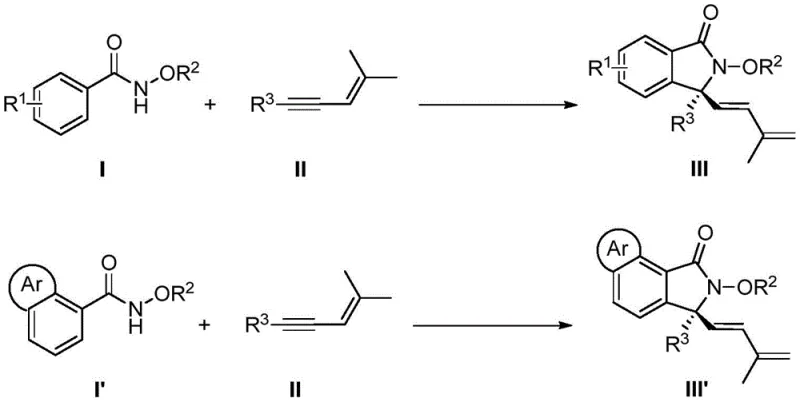
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to construct complex chiral scaffolds, particularly those containing quaternary carbon centers which are prevalent in bioactive molecules. Patent CN113735756A introduces a groundbreaking methodology for the synthesis of chiral 3,3-disubstituted isoindolinone compounds, utilizing an easily prepared chiral cyclopentadienyl rhodium catalyst. This innovation represents a significant leap forward in asymmetric C-H bond activation, enabling the direct coupling of N-methoxybenzamide derivatives with 1,3-enyne compounds. The process achieves high yields and exceptional enantioselectivity under remarkably mild reaction conditions, typically between 5°C and 15°C. By streamlining the synthetic route through a tandem sequence involving C-H activation, enyne migration insertion, 1,4-rhodium migration, and nucleophilic cyclization, this technology overcomes the historical limitations of multi-step syntheses that often suffer from poor atom economy and difficult substrate preparation. For a reliable pharmaceutical intermediate supplier, mastering such direct functionalization techniques is crucial for maintaining competitiveness in the global market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the construction of 3,3-disubstituted chiral isoindolinone skeletons has been a formidable challenge for organic chemists due to the steric congestion around the quaternary carbon center. Conventional strategies often rely on the enantioselective functionalization of pre-existing isoindolinone cores, which necessitates the prior synthesis of the lactam ring itself. This multi-step approach inherently suffers from low atom and step economy, as each additional transformation introduces potential yield losses and generates chemical waste. Furthermore, the starting materials required for these traditional routes are frequently unstable or difficult to prepare, limiting the structural diversity of the final products. The harsh reaction conditions often employed in classical methods, such as strong bases or high temperatures, can lead to racemization of the chiral center or decomposition of sensitive functional groups. These factors collectively result in higher production costs and longer lead times, creating significant bottlenecks for procurement managers seeking cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
In stark contrast, the novel approach detailed in the patent leverages transition metal-catalyzed direct C-H bond functionalization to construct the target skeleton in a single pot. By employing N-methoxybenzamides and 1,3-enynes as simple, stable, and readily available starting materials, the method bypasses the need for pre-activated substrates. The use of a chiral cyclopentadienyl rhodium catalyst allows for precise stereocontrol during the [4+1] cyclization event, effectively building the quaternary center with high fidelity. This strategy not only simplifies the operational procedure but also drastically improves the overall efficiency of the synthesis. The ability to tolerate a wide range of functional groups on the benzamide ring, including halogens, alkyls, and electron-withdrawing groups, demonstrates the robustness of this catalytic system. For supply chain heads, this translates to reduced dependency on complex precursor supply chains and enhanced flexibility in sourcing raw materials for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Rhodium-Catalyzed Enantioselective [4+1] Cyclization
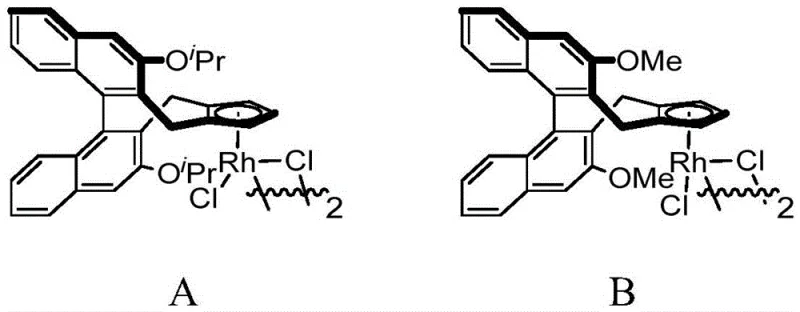
The core of this technological breakthrough lies in the sophisticated design of the chiral catalyst and the intricate mechanistic pathway it facilitates. The reaction initiates with the coordination of the rhodium center to the directing group of the N-methoxybenzamide, followed by the cleavage of the ortho C-H bond to form a rhodacycle intermediate. This step is critical as it defines the regioselectivity of the subsequent transformation. The 1,3-enyne then undergoes migratory insertion into the Rh-C bond, a process that is highly sensitive to the steric environment created by the chiral cyclopentadienyl ligand. The specific architecture of the ligand, featuring bulky substituents like isopropoxy or methoxy groups on the binaphthyl backbone, creates a chiral pocket that discriminates between the enantiotopic faces of the incoming alkyne. Following insertion, a rare 1,4-rhodium migration occurs, shifting the metal center to a position conducive for the final ring-closing step. This migration is key to forming the five-membered isoindolinone ring with the correct connectivity.
Furthermore, the mechanism ensures rigorous impurity control through the high specificity of the catalytic cycle. The use of silver difluoride as an oxidant facilitates the regeneration of the active Rh(III) species while minimizing side reactions that could lead to byproduct formation. The mild acidic conditions provided by the carboxylic acid additive help to protonate the intermediate at the correct stage, promoting nucleophilic attack by the amide oxygen to close the ring. This precise control over the reaction trajectory prevents the formation of regioisomers or oligomeric byproducts that are common in less selective alkyne coupling reactions. For R&D directors, understanding these mechanistic nuances is vital for troubleshooting and optimizing the process for specific API intermediates. The high enantiomeric excess (ee) values reported, reaching up to 96% in optimized examples, indicate that the chiral information is effectively transferred from the catalyst to the product, reducing the need for costly downstream chiral resolution processes.
How to Synthesize Chiral 3,3-Disubstituted Isoindolinone Efficiently
Implementing this synthesis requires careful attention to reaction parameters to maximize both yield and stereoselectivity. The protocol involves dissolving the N-methoxybenzamide substrate and the 1,3-enyne coupling partner in an alcohol solvent, such as 3-pentanol or ethanol, which plays a dual role as a medium and potentially as a proton source. The addition of the chiral rhodium catalyst, typically at a loading of 3-5 mol%, along with the silver oxidant and acetic acid additive, must be performed under controlled conditions to ensure homogeneity. The reaction is then maintained at a low temperature range of 5-15°C for an extended period of 60 to 80 hours. This prolonged reaction time at low temperature is essential to allow the slow, selective catalytic cycles to proceed without triggering non-selective background reactions. Detailed standardized synthesis steps see the guide below.
- Prepare the reaction mixture by combining N-methoxybenzamide, 1,3-enyne compound, chiral cyclopentadienyl rhodium catalyst, oxidant, and carboxylic acid additive in an alcohol solvent.
- Maintain the reaction temperature between 5-15°C and stir continuously for a duration of 60 to 80 hours to ensure complete conversion and high stereoselectivity.
- Quench the reaction with ethylenediamine, remove the solvent, and purify the crude product via silica gel column chromatography to isolate the target chiral isoindolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this rhodium-catalyzed methodology offers substantial benefits that directly impact the bottom line and operational resilience of chemical manufacturing enterprises. The shift from multi-step batch processes to a more convergent, one-pot synthesis significantly reduces the total processing time and the number of unit operations required. This consolidation of steps leads to a drastic simplification of the manufacturing workflow, lowering labor costs and minimizing the footprint of the production facility. Moreover, the use of stable and commercially available starting materials mitigates the risk of supply chain disruptions associated with exotic or custom-synthesized reagents. The mild reaction conditions also imply lower energy consumption for heating or cooling, contributing to a more sustainable and cost-effective production profile. For procurement managers, these factors combine to offer a compelling value proposition for sourcing high-purity intermediates.
- Cost Reduction in Manufacturing: The elimination of pre-functionalization steps and the high atom economy of the [4+1] cyclization directly translate to reduced raw material consumption. By avoiding the use of expensive protecting groups and harsh reagents, the overall cost of goods sold (COGS) is significantly lowered. Additionally, the high selectivity of the catalyst minimizes the formation of difficult-to-separate impurities, which reduces the burden on purification units and increases the overall recovery of the desired product. This efficiency gain allows for competitive pricing strategies in the global market for fine chemicals.
- Enhanced Supply Chain Reliability: The robustness of the reaction towards various functional groups means that a single platform technology can be applied to synthesize a diverse library of analogues. This versatility reduces the need for developing entirely new processes for each new derivative, accelerating time-to-market for new drug candidates. The stability of the N-methoxybenzamide precursors ensures long shelf-life and ease of storage, facilitating better inventory management and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process operates in alcohol solvents, which are generally greener and easier to recover than chlorinated or aromatic solvents often used in traditional heterocycle synthesis. The low catalyst loading and the potential for catalyst recycling further enhance the environmental profile of the process. Scaling this reaction from gram to kilogram or ton scale is facilitated by the mild thermal requirements, reducing the safety risks associated with exothermic runaway reactions. This aligns well with increasingly stringent environmental regulations and corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this rhodium-catalyzed synthesis technology. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation. Understanding these details helps stakeholders evaluate the feasibility of integrating this method into their existing production pipelines. The answers reflect the current state of the art in asymmetric C-H activation and provide a realistic overview of the process capabilities.
Q: What are the key advantages of this rhodium-catalyzed method over traditional synthesis?
A: This method utilizes a direct C-H bond activation strategy, eliminating the need for pre-functionalized substrates and multi-step sequences. It operates under exceptionally mild conditions (5-15°C) and achieves high enantioselectivity (up to 96% ee), significantly reducing energy consumption and purification costs compared to conventional harsh protocols.
Q: Which catalysts and oxidants are preferred for optimal yield and selectivity?
A: The patent specifies chiral cyclopentadienyl rhodium complexes (specifically Catalyst A or B with methoxy or isopropoxy groups) as the core catalysts. Silver difluoride (AgF2) is identified as the preferred oxidant, while acetic acid serves as the optimal carboxylic acid additive to facilitate the catalytic cycle and proton transfer steps.
Q: Is this process suitable for large-scale industrial production of pharmaceutical intermediates?
A: Yes, the process demonstrates excellent substrate scope and operational simplicity. The use of stable, commercially available starting materials like N-methoxybenzamides and 1,3-enynes, combined with low catalyst loading (3-5 mol%), makes it highly viable for commercial scale-up, addressing supply chain continuity for complex chiral scaffolds.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 3,3-Disubstituted Isoindolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic technologies like the one described in CN113735756A for the next generation of pharmaceutical intermediates. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that laboratory breakthroughs are seamlessly translated into industrial reality. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of complex chiral scaffolds or large-scale supply of key building blocks, our infrastructure is designed to meet the demanding quality standards of the global pharmaceutical industry.
We invite you to collaborate with us to explore how this efficient rhodium-catalyzed route can optimize your supply chain and reduce your manufacturing costs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to support your R&D and commercialization efforts. Let us be your partner in navigating the complexities of modern chemical synthesis.