Scalable Synthesis of 2-Trifluoromethyl Imidazoles via Mild Palladium-Catalyzed Carbonylation for Advanced Drug Discovery
Scalable Synthesis of 2-Trifluoromethyl Imidazoles via Mild Palladium-Catalyzed Carbonylation for Advanced Drug Discovery
The integration of trifluoromethyl groups into heterocyclic scaffolds represents a cornerstone strategy in modern medicinal chemistry, significantly enhancing the metabolic stability, lipophilicity, and bioavailability of drug candidates. As highlighted in recent patent literature, specifically CN111423381B, there is a critical demand for efficient methodologies to access these privileged structures without resorting to harsh reaction conditions or exotic reagents. This patent discloses a robust preparation method for 2-trifluoromethyl substituted imidazole compounds, leveraging a transition metal palladium-catalyzed carbonylation series reaction. The significance of this technology lies in its ability to operate at a remarkably mild temperature of 30°C, utilizing inexpensive starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts. For research and development teams seeking reliable pharmaceutical intermediate suppliers, this approach offers a pathway to complex nitrogen-containing five-membered heterocycles that are ubiquitous in bioactive molecules, ranging from histamine receptor antagonists to sophisticated NHC ligands used in coordination catalysis.
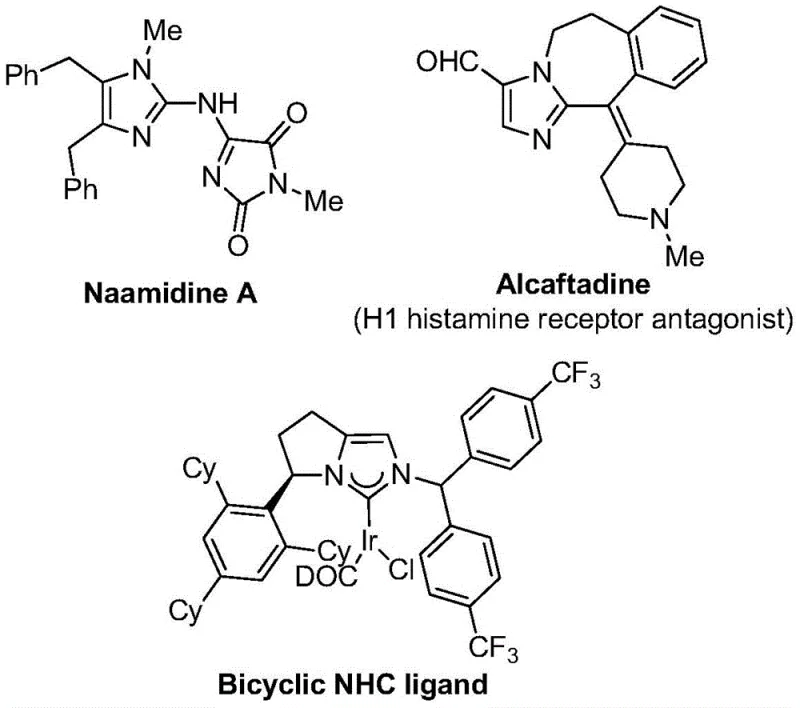
The structural diversity achievable through this synthetic route is substantial, allowing for the precise installation of trifluoromethyl groups alongside various aryl substitutions. This capability is paramount for optimizing the physicochemical properties of lead compounds during the drug discovery phase. By employing a carbonylation strategy that generates carbon monoxide in situ from formic acid and acetic anhydride, the method circumvents the safety hazards associated with handling high-pressure CO gas. Furthermore, the compatibility with a wide array of functional groups ensures that late-stage functionalization is feasible, thereby accelerating the timeline for generating analog libraries. This technological advancement positions the synthesis of 2-trifluoromethyl imidazoles not merely as an academic exercise but as a practical solution for cost reduction in API manufacturing, where efficiency and safety are non-negotiable metrics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted nitrogen-containing heterocycles has been plagued by significant operational challenges and limited substrate scope. Traditional methods often rely on the direct reaction of synthons bearing trifluoromethyl groups, such as trifluorodiazoethane, with suitable substrates under rigorous conditions. These conventional pathways frequently necessitate elevated temperatures, strong bases, or hazardous reagents that pose severe safety risks in a production environment. Moreover, the availability of specialized trifluoromethyl synthons has been a bottleneck; many are expensive, unstable, or difficult to procure in bulk quantities, leading to supply chain vulnerabilities. The reliance on such reagents often results in poor atom economy and generates substantial chemical waste, complicating downstream purification and increasing the environmental footprint of the manufacturing process. Additionally, older methodologies often struggle with regioselectivity, producing mixtures of isomers that require tedious separation techniques, ultimately driving up the cost of goods and extending lead times for high-purity pharmaceutical intermediates.
The Novel Approach
In stark contrast, the novel approach detailed in patent CN111423381B revolutionizes the landscape by utilizing a palladium-catalyzed multicomponent coupling reaction that is both operationally simple and highly efficient. This method employs cheap and easily obtained starting materials, specifically trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts, which are commercially accessible and stable. The reaction proceeds under exceptionally mild conditions at 30°C, drastically reducing energy consumption compared to thermal-intensive legacy processes. The use of a formic acid/acetic anhydride system as a safe carbon monoxide surrogate eliminates the need for specialized high-pressure equipment, making the process inherently safer and more adaptable to standard reactor setups. This innovation facilitates the commercial scale-up of complex pharmaceutical intermediates by ensuring high reaction efficiency and excellent substrate compatibility. The ability to synthesize diversely substituted imidazole compounds through simple substrate design broadens the utility of this method, allowing chemists to rapidly explore chemical space without being constrained by the limitations of traditional trifluoromethylation reagents.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The mechanistic pathway of this transformation is a sophisticated orchestration of organometallic steps that ensures high fidelity and yield. The reaction initiates with the formation of an intermolecular carbon-nitrogen bond promoted by the alkali additive, yielding a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization, setting the stage for the palladium cycle. The palladium catalyst, generated in situ from PdCl2 and triphenylphosphine, coordinates with the alkyne moiety of the propargylamine derivative, facilitating a palladation event to form a key alkenyl palladium intermediate. This intermediate then isomerizes to a more stable alkyl palladium species, which is primed for the crucial carbonylation step. Under the influence of carbon monoxide released from the decomposition of the formic acid/acetic anhydride mixture, an acyl palladium intermediate is generated. This acyl species is highly reactive and poised for the final bond-forming events that construct the imidazole core.
The catalytic cycle culminates in the oxidative addition of the diaryl iodonium salt to the palladium center, forming a transient tetravalent palladium intermediate. This high-valent species is unstable and rapidly undergoes reductive elimination, forging the final carbon-carbon bond and releasing the desired 2-trifluoromethyl-substituted imidazole product while regenerating the active palladium catalyst. Understanding this mechanism is vital for process optimization, as it highlights the critical role of each component: the base facilitates the initial amidine formation, the CO source drives the carbonylation, and the iodonium salt acts as the arylating agent. From an impurity control perspective, the mild conditions minimize side reactions such as polymerization of the alkyne or decomposition of the sensitive imidoyl chloride. The specificity of the palladium cycle ensures that the trifluoromethyl group remains intact throughout the process, preserving the electronic properties essential for the biological activity of the final molecule. This mechanistic clarity provides a solid foundation for scaling the reaction while maintaining strict quality control standards required for high-purity OLED material or pharmaceutical applications.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
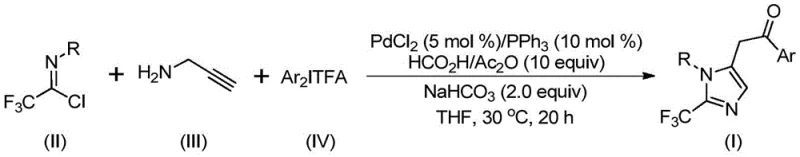
Executing this synthesis requires careful attention to reagent stoichiometry and reaction monitoring to maximize yield and purity. The protocol outlined in the patent suggests a molar ratio of palladium chloride to triphenylphosphine to sodium bicarbonate of approximately 0.05:0.1:2, ensuring sufficient catalytic activity and neutralization of acidic byproducts. The reaction is typically conducted in tetrahydrofuran (THF), which effectively dissolves all reactants and promotes the catalytic cycle. While the standard procedure involves reacting the mixture for 20 hours, monitoring conversion is recommended to determine the optimal endpoint for specific substrate combinations. The post-treatment is straightforward, involving filtration to remove inorganic salts followed by silica gel mixing and column chromatography purification.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, formic acid, trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt in an organic solvent such as THF.
- Stir the reaction mixture at a mild temperature of 30°C for a duration of 16 to 24 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target 2-trifluoromethyl substituted imidazole compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic advantages that extend beyond mere chemical elegance. The primary driver for cost efficiency is the utilization of commodity chemicals as starting materials. Trifluoroethylimidoyl chloride can be rapidly synthesized from inexpensive aromatic amines, while propargylamine and diaryl iodonium salts are widely available in the global chemical market. This reliance on abundant feedstocks mitigates the risk of supply disruptions that often plague processes dependent on proprietary or niche reagents. Furthermore, the elimination of high-pressure carbon monoxide cylinders reduces the capital expenditure required for specialized reactor infrastructure and lowers the regulatory burden associated with handling toxic gases. These factors collectively contribute to a leaner, more resilient supply chain capable of responding swiftly to market demands for reliable pharmaceutical intermediate suppliers.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the low loading of the palladium catalyst (5 mol%) and the use of triphenylphosphine, which are relatively inexpensive compared to specialized ligands often required in cross-coupling reactions. The mild reaction temperature of 30°C translates to significant energy savings, as there is no need for extensive heating or cooling systems to maintain thermal equilibrium. Additionally, the high atom economy of the carbonylation cyclization minimizes waste generation, reducing the costs associated with waste disposal and environmental compliance. By streamlining the synthetic sequence into a one-pot operation, labor costs and processing time are substantially reduced, leading to a lower overall cost of goods sold without compromising on the quality of the final product.
- Enhanced Supply Chain Reliability: The robustness of this methodology against variations in substrate structure ensures consistent production outcomes, which is critical for maintaining uninterrupted supply lines. The tolerance for diverse functional groups means that a single standardized protocol can be applied to manufacture a wide library of derivatives, simplifying inventory management and production planning. Since the key reagents are stable and commercially sourced, the lead time for raw material acquisition is minimized, allowing for just-in-time manufacturing strategies. This reliability is essential for partners seeking to secure long-term contracts for commercial scale-up of complex pharmaceutical intermediates, as it guarantees that production schedules can be met even in volatile market conditions.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram or ton scale is facilitated by the absence of hazardous high-pressure steps and the use of common organic solvents like THF. The simple workup procedure, which avoids complex extractions or crystallizations in favor of standard chromatography or filtration, is easily adaptable to continuous flow chemistry or large batch reactors. From an environmental standpoint, the generation of carbon monoxide in situ from formic acid is a greener alternative to using fossil-fuel-derived CO, aligning with modern sustainability goals. The reduced formation of byproducts and the high selectivity of the reaction lower the E-factor (environmental factor), making this process attractive for companies aiming to reduce their carbon footprint and adhere to stringent environmental regulations governing chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this palladium-catalyzed synthesis. These insights are derived directly from the experimental data and scope analysis presented in the patent documentation, providing a clear understanding of the method's capabilities and limitations. Addressing these points helps stakeholders evaluate the feasibility of integrating this technology into their existing manufacturing workflows.
Q: What are the optimal reaction conditions for this palladium-catalyzed synthesis?
A: The patent specifies using PdCl2 (5 mol%) and PPh3 (10 mol%) in THF solvent with NaHCO3 as a base. The reaction proceeds efficiently at a mild 30°C over 20 hours, utilizing formic acid and acetic anhydride as the carbon monoxide source.
Q: Does this method tolerate diverse functional groups on the substrate?
A: Yes, the methodology demonstrates excellent substrate compatibility. It successfully accommodates various substituents including methyl, tert-butyl, methoxy, chloro, bromo, trifluoromethyl, and nitro groups on both the imidoyl chloride and the diaryl iodonium salt components.
Q: Is this process suitable for large-scale industrial production?
A: The process is designed for scalability, utilizing cheap and readily available starting materials like propargylamine and aromatic amines. The mild reaction temperature and simple post-treatment involving filtration and chromatography make it highly viable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the pivotal role that advanced heterocyclic intermediates play in the development of next-generation therapeutics and functional materials. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether your project requires custom synthesis of novel trifluoromethyl imidazoles or the reliable supply of established intermediates, our infrastructure is designed to support your growth and innovation.
We invite you to engage with our technical procurement team to discuss how this patented technology can be leveraged for your specific applications. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how optimizing your synthetic route can improve margins and efficiency. We encourage potential partners to contact us for specific COA data and route feasibility assessments, ensuring that every batch delivered meets the highest standards of quality and performance required by the global pharmaceutical and fine chemical industries.