Advanced Synthetic Routes for High-Purity Entecavir Manufacturing and Commercial Scale-Up
Advanced Synthetic Routes for High-Purity Entecavir Manufacturing and Commercial Scale-Up
The pharmaceutical industry continuously seeks robust and scalable methodologies for the production of critical antiviral agents, particularly nucleoside analogues like Entecavir. Patent CN101130542B introduces a transformative synthesis method that addresses long-standing inefficiencies in traditional manufacturing pathways. This technology shifts the paradigm from direct guanine ring-opening to a strategic coupling of methylene cyclopentane amines with pyrimidine derivatives. 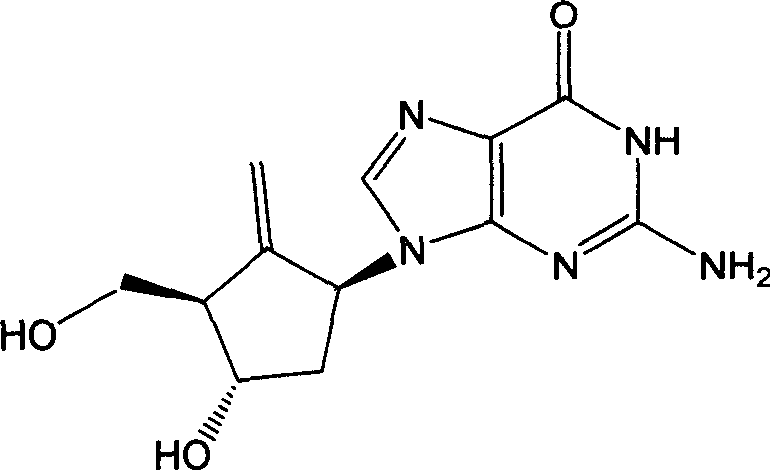 As depicted in the structural analysis, Entecavir is a potent carbocyclic guanosine analogue used extensively for treating hepatitis B virus infections. The innovation lies not just in the final molecule, but in the architectural redesign of the synthetic route to enhance regioselectivity and simplify purification, offering a compelling value proposition for reliable pharmaceutical intermediate suppliers aiming to optimize their production lines.
As depicted in the structural analysis, Entecavir is a potent carbocyclic guanosine analogue used extensively for treating hepatitis B virus infections. The innovation lies not just in the final molecule, but in the architectural redesign of the synthetic route to enhance regioselectivity and simplify purification, offering a compelling value proposition for reliable pharmaceutical intermediate suppliers aiming to optimize their production lines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of carbocyclic nucleosides relied heavily on the direct ring-opening of epoxidized cyclopentane compounds using guanine derivatives, as documented in prior art such as WO9809964 and JOC1985. This conventional approach suffers from severe intrinsic drawbacks that hinder industrial viability. Primarily, the nucleophilic attack on the guanine ring lacks perfect regioselectivity, resulting in a mixture of 9-N and 7-N isomers that are notoriously difficult to separate even after multiple silica gel column chromatography steps. Furthermore, the yield of this ring-opening reaction is often unacceptably low, reported around 27% to 50% in various literature sources. The necessity to protect the 2-amino group on the guanine moiety adds further complexity, as protecting groups like MMT are unstable and require harsh conditions for removal, often leading to product decomposition and necessitating specialized resin chromatography for final purification.
The Novel Approach
In stark contrast, the methodology disclosed in CN101130542B circumvents these bottlenecks by reversing the construction logic. Instead of attaching a pre-formed guanine base to the sugar mimic, the process couples a functionalized methylene cyclopentane amine with a pyrimidine derivative carrying a leaving group at the 6-position. 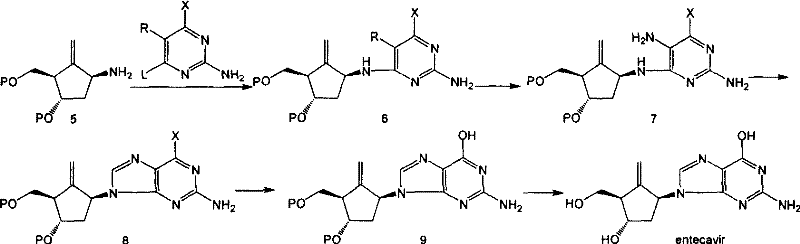 This strategic pivot ensures that the nitrogen atom connecting the base to the cyclopentane ring is predetermined, effectively eliminating the formation of unwanted 7-N regioisomers. By deferring the formation of the purine ring until after the coupling step, the synthesis avoids the cumbersome protection and deprotection cycles associated with the guanine 2-amino group. This results in a streamlined workflow where intermediates are more stable, separation is simplified, and the overall process becomes far more amenable to cost reduction in pharmaceutical intermediates manufacturing.
This strategic pivot ensures that the nitrogen atom connecting the base to the cyclopentane ring is predetermined, effectively eliminating the formation of unwanted 7-N regioisomers. By deferring the formation of the purine ring until after the coupling step, the synthesis avoids the cumbersome protection and deprotection cycles associated with the guanine 2-amino group. This results in a streamlined workflow where intermediates are more stable, separation is simplified, and the overall process becomes far more amenable to cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Pyrimidine-Purine Cyclization Strategy
The core mechanistic advantage of this novel route lies in the controlled assembly of the heterocyclic system. The process begins with a nucleophilic substitution where the primary amine of the methylene cyclopentane attacks the electron-deficient carbon at the 6-position of the pyrimidine ring, displacing a leaving group such as chlorine. This step is typically conducted in polar solvents like n-butanol or DMF with an organic amine base to scavenge the generated acid. Following the coupling, the critical ring-closing step transforms the pyrimidine intermediate into the target purine system. This cyclization is catalyzed by strong acids in the presence of trialkyl orthoformates, which serve as both solvent and one-carbon source for closing the imidazole ring. The use of specific precursors, such as 2-amino-5-nitro-4-chloropyrimidine, allows for subsequent reduction of the nitro group to an amine, facilitating the cyclization under mild conditions. 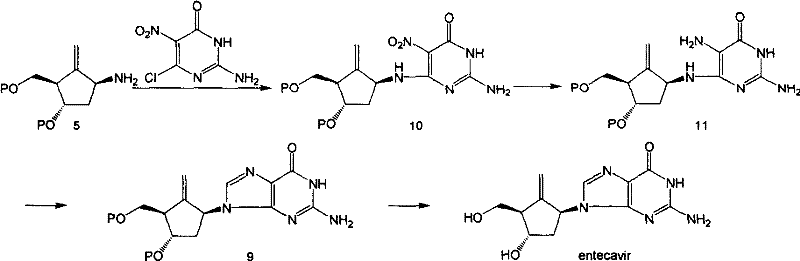 This mechanistic pathway ensures high conversion rates and minimizes side reactions, providing a clean impurity profile that is essential for meeting stringent regulatory standards in API production.
This mechanistic pathway ensures high conversion rates and minimizes side reactions, providing a clean impurity profile that is essential for meeting stringent regulatory standards in API production.
Furthermore, the final deprotection strategy employed in this patent demonstrates superior chemoselectivity compared to traditional silane-based methods. Conventional routes often utilize silyl protecting groups that require aggressive oxidative or acidic conditions for removal, which can degrade the sensitive exocyclic double bond of the cyclopentane ring. The disclosed method utilizes benzyl protecting groups that can be cleanly removed using boron trichloride at low temperatures, typically around -78°C to -20°C. This mild deprotection preserves the integrity of the methylene group and the stereochemistry of the chiral centers. The ability to perform these transformations without resorting to special resin chromatography indicates a significant advancement in process chemistry, allowing for the isolation of high-purity Entecavir through standard crystallization techniques, thereby enhancing the commercial scale-up of complex nucleoside analogues.
How to Synthesize Entecavir Efficiently
The synthesis of Entecavir via this patented route involves a sequence of well-defined chemical transformations designed for reproducibility and scalability. The process initiates with the preparation of the key amino-cyclopentane intermediate, followed by coupling with a substituted pyrimidine, cyclization to form the purine core, and final deprotection. Each step has been optimized to maximize yield while minimizing the generation of hazardous waste or difficult-to-remove impurities. For R&D teams looking to implement this technology, the detailed standardized synthesis steps are provided in the guide below, ensuring that the transition from laboratory bench to pilot plant is seamless and compliant with good manufacturing practices.
- Couple methylene cyclopentane amine with a 6-position substituted pyrimidine derivative in polar solvent with an acid-binding agent.
- Perform ring-closing reaction using trialkyl orthoformate and strong acid catalysis to form the purine skeleton.
- Execute final deprotection using boron trichloride or hydrolysis to remove protecting groups and yield pure Entecavir.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical elegance. The elimination of low-yielding ring-opening steps and the removal of complex chromatographic purification requirements directly translate to substantial cost savings in raw material utilization and processing time. By avoiding the use of unstable protecting groups and harsh deprotection conditions, the process reduces the risk of batch failures and rework, ensuring a more consistent supply of high-quality intermediates. This reliability is crucial for maintaining continuous production schedules in the competitive antiviral market, where demand fluctuations can be sharp and unpredictable.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis eliminates the need for expensive and time-consuming column chromatography steps that are prevalent in older methods. By relying on crystallization and standard extraction techniques for purification, manufacturers can drastically reduce solvent consumption and labor costs associated with silica gel handling. Additionally, the higher overall yield means less starting material is required to produce the same amount of final API, leading to significant optimization of the cost of goods sold without compromising on quality standards.
- Enhanced Supply Chain Reliability: The reagents utilized in this pathway, such as substituted pyrimidines and trialkyl orthoformates, are commercially available commodity chemicals with stable supply chains. Unlike specialized chiral catalysts or sensitive organometallic reagents that might face sourcing bottlenecks, these materials ensure that production is not held hostage by single-source supplier issues. The robustness of the reaction conditions, which tolerate standard industrial equipment and do not require exotic pressure or temperature controls, further secures the supply continuity for high-purity nucleoside analogues.
- Scalability and Environmental Compliance: From an environmental and safety perspective, this route is inherently greener due to the reduction in solvent waste and the avoidance of heavy metal catalysts often found in alternative cross-coupling methods. The ability to scale this process from kilogram to multi-ton quantities is supported by the use of common solvents like n-butanol and ethanol, which are easier to recover and recycle than chlorinated solvents. This alignment with green chemistry principles not only lowers waste disposal costs but also facilitates smoother regulatory approvals in regions with strict environmental compliance mandates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Entecavir synthesis technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, providing a clear understanding of the operational benefits. Understanding these nuances is vital for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: How does this new synthesis route improve upon conventional guanine ring-opening methods?
A: The novel route avoids the low-yield and difficult-to-separate stereoisomers associated with direct guanine ring-opening. By coupling an amino-cyclopentane with a pyrimidine derivative first, regioselectivity is inherently controlled, eliminating the need for complex chromatographic separation of 7-N and 9-N isomers.
Q: What are the key advantages regarding impurity control in this process?
A: This method significantly reduces impurity profiles by avoiding the protection and deprotection of the guanine 2-amino group, which is prone to instability. The intermediates formed are more stable and easier to purify, leading to a final product with higher pharmaceutical-grade purity without resin chromatography.
Q: Is this synthetic pathway suitable for large-scale industrial production?
A: Yes, the process utilizes robust reaction conditions such as reflux in common polar solvents and standard reagents like sodium dithionite and triethyl orthoformate. The elimination of sensitive protection groups and difficult column chromatography steps makes it highly scalable for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Entecavir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the modern pharmaceutical landscape. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical advantages of patents like CN101130542B are fully realized in practical manufacturing settings. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Entecavir intermediate meets the highest global pharmacopoeia standards, providing our partners with the confidence needed for successful drug development and commercialization.
We invite global pharmaceutical companies and contract manufacturers to collaborate with us to leverage this advanced technology for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, ensuring that your project benefits from the most efficient and economically viable synthesis strategy available in the market.