Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Drug Manufacturing
Introduction to Next-Generation Quinazolinone Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for more efficient and cost-effective synthetic routes for privileged scaffolds. A groundbreaking development detailed in patent CN113045503B introduces a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds, a structural motif prevalent in numerous bioactive molecules. This innovation addresses critical bottlenecks in the production of drugs such as Methaqualone and the natural alkaloid Rutaecarpine, which are known for their antifungal, antiviral, and anticancer properties. By leveraging a transition metal palladium-catalyzed carbonylation cascade, this technology enables the direct assembly of the quinazolinone core from simple, commercially available starting materials.
The strategic introduction of the trifluoromethyl group into heterocyclic systems is a key focus for modern drug design, as it significantly enhances metabolic stability, lipophilicity, and bioavailability. However, traditional synthetic pathways often struggle to incorporate this group efficiently without compromising yield or requiring hazardous reagents. The method disclosed in this patent overcomes these limitations by utilizing trifluoroethylimidoyl chloride as a robust building block. This approach not only simplifies the operational workflow but also expands the chemical space accessible to process chemists, offering a reliable pathway for the commercial scale-up of complex pharmaceutical intermediates.
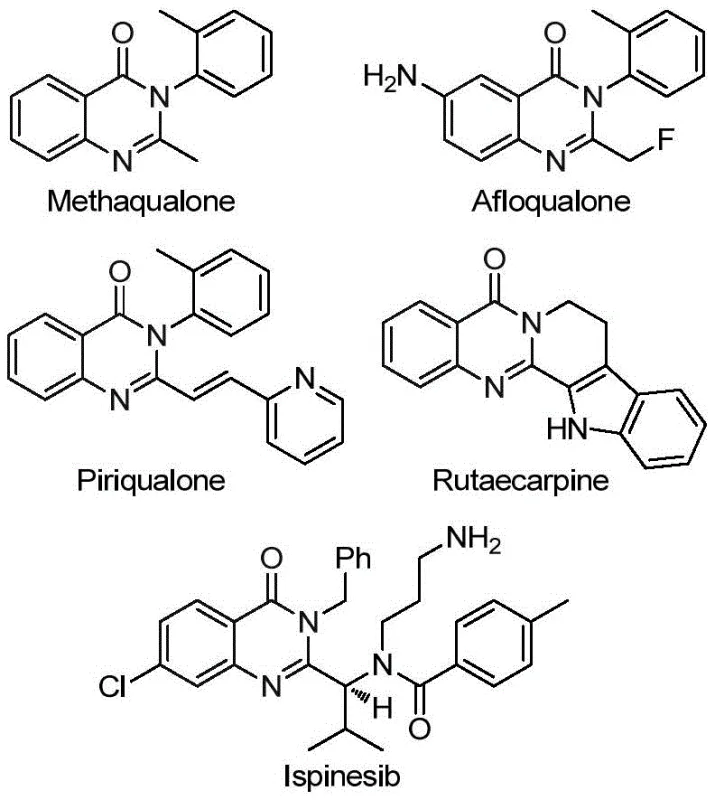
For R&D directors and procurement specialists, understanding the mechanistic underpinnings and practical advantages of this synthesis is crucial for supply chain optimization. The ability to synthesize these compounds with high purity and minimal waste translates directly into reduced manufacturing costs and improved supply continuity. As we delve deeper into the technical specifics, it becomes evident that this methodology represents a significant leap forward in the cost reduction in pharmaceutical intermediate manufacturing, providing a competitive edge for companies looking to streamline their production of fluorinated heterocycles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl-substituted quinazolinones has been plagued by significant technical hurdles that hinder large-scale production. Conventional literature reports primarily rely on the cyclization of anthranilamide with ethyl trifluoroacetate, trifluoroacetic anhydride, or trifluoroacetic acid under varying conditions. Alternatively, methods involving the reaction of anthranilates with unstable trifluoroacetamides or the cyclization of isatoic anhydride with trifluoroacetic anhydride are common. These legacy processes suffer from severe drawbacks, including harsh reaction conditions that demand precise temperature control and specialized equipment, increasing operational risks and energy consumption.
Furthermore, the substrates required for these traditional routes, such as isatoic anhydride or pre-activated trifluoroacetamides, are often expensive and not readily available in bulk quantities, creating supply chain vulnerabilities. The reaction efficiencies are frequently suboptimal, with reported yields often falling short of industrial expectations, leading to significant material loss and increased waste generation. Additionally, the substrate scope is typically narrow, limiting the ability to introduce diverse functional groups necessary for structure-activity relationship (SAR) studies. These factors collectively result in a high cost of goods sold (COGS) and extended lead times, making conventional methods less attractive for reliable pharmaceutical intermediate supplier networks aiming for agility and cost-efficiency.
The Novel Approach
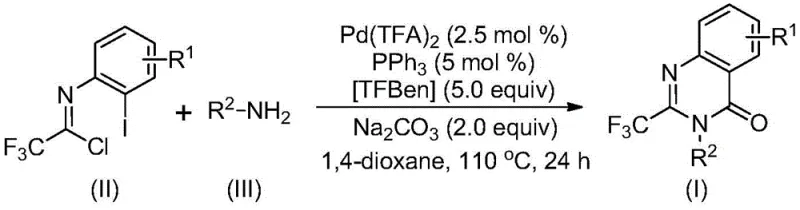
In stark contrast, the novel methodology presented in the patent utilizes a palladium-catalyzed carbonylation cascade reaction that fundamentally reshapes the synthetic landscape for these compounds. By employing cheap and easily accessible trifluoroethylimidoyl chloride and various amines as starting materials, the process eliminates the need for expensive pre-activation steps. The reaction proceeds smoothly in the presence of a palladium catalyst, a phosphine ligand, and a carbon monoxide substitute (TFBen), facilitating the formation of the quinazolinone ring under relatively mild thermal conditions. This shift in strategy allows for the direct construction of the core scaffold with exceptional efficiency and atom economy.
The versatility of this new approach is demonstrated by its broad substrate compatibility, accommodating a wide array of amines including alkyl, cycloalkyl, benzyl, and substituted aryl amines. The reaction tolerates various functional groups on the aromatic ring, such as halogens and electron-donating or withdrawing groups, enabling the rapid generation of diverse libraries for drug discovery. Moreover, the operational simplicity—mixing reagents in a solvent like 1,4-dioxane and heating—makes it highly amenable to scale-up. This robustness ensures consistent quality and yield, addressing the critical need for high-purity pharmaceutical intermediates while drastically simplifying the manufacturing workflow compared to legacy techniques.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The success of this synthesis lies in the intricate catalytic cycle driven by the palladium complex. The reaction likely initiates with a base-promoted intermolecular carbon-nitrogen bond coupling between the amine and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine derivative in situ. Subsequently, the palladium catalyst inserts into the carbon-iodine bond of the aromatic ring, forming a reactive divalent palladium intermediate. This step is critical as it activates the aryl ring for the subsequent carbonylation event, setting the stage for ring closure.
Under the heating conditions employed, the additive TFBen (1,3,5-tricarboxylic acid phenol ester) decomposes to release carbon monoxide, which serves as the carbonyl source. This CO molecule inserts into the carbon-palladium bond to form an acyl palladium intermediate. The presence of the base then facilitates the coordination of the nitrogen atom to the palladium center, promoting the formation of a seven-membered ring palladium intermediate. Finally, a reductive elimination step occurs, releasing the final 2-trifluoromethyl-substituted quinazolinone product and regenerating the active palladium catalyst. This elegant mechanism ensures high turnover and minimizes side reactions, contributing to the observed high yields.
From an impurity control perspective, the choice of ligands and the specific ratio of catalyst to substrate play a pivotal role. The use of triphenylphosphine as a ligand stabilizes the palladium species, preventing aggregation and deactivation, which are common sources of metallic impurities in final products. The controlled release of CO from TFBen avoids the safety hazards associated with handling gaseous carbon monoxide directly, while ensuring a steady concentration of the carbonyl source to drive the reaction to completion. This precise control over the reaction environment results in a clean crude profile, simplifying downstream purification and ensuring that the final high-purity pharmaceutical intermediates meet stringent regulatory specifications for residual metals and organic impurities.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
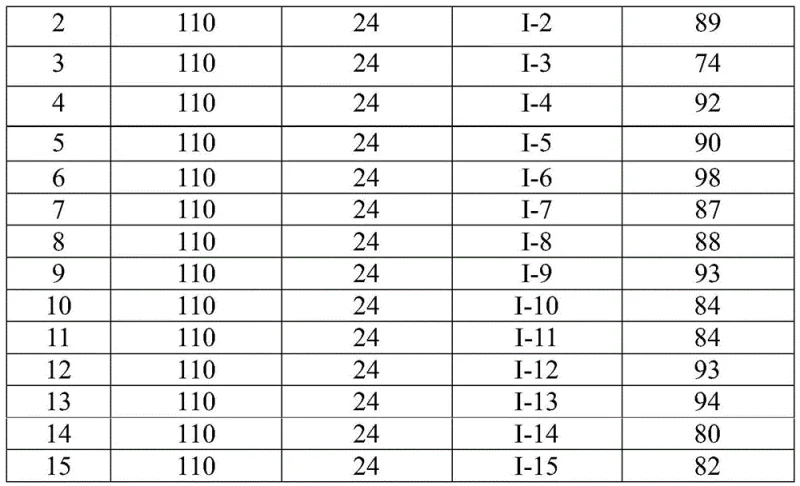
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific protocols to maximize yield and safety. The process begins with the careful weighing and mixing of the palladium catalyst, ligand, base, and the unique CO surrogate TFBen along with the substrates in an anhydrous organic solvent. The reaction is then heated to a specific temperature range to activate the catalytic cycle. Following the reaction period, a straightforward workup involving filtration and chromatography yields the target compound. For detailed operational parameters and safety guidelines, please refer to the standardized synthesis steps provided below.
- Combine palladium trifluoroacetate, triphenylphosphine, TFBen, sodium carbonate, trifluoroethylimidoyl chloride, and amine in an organic solvent such as 1,4-dioxane.
- Heat the reaction mixture to 110°C and maintain stirring for 16 to 30 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers transformative benefits that extend beyond mere chemical efficiency. The primary advantage lies in the substantial reduction of raw material costs. By replacing expensive and unstable precursors like isatoic anhydride or pre-activated trifluoroacetamides with cheap, commercially available trifluoroethylimidoyl chloride and simple amines, the direct material cost is significantly lowered. This shift not only improves the margin profile of the final product but also mitigates the risk of price volatility associated with specialty reagents, ensuring a more stable and predictable cost structure for long-term contracts.
Furthermore, the enhanced supply chain reliability is a direct consequence of the method's reliance on commodity chemicals. The starting materials are widely produced and stocked by multiple global suppliers, eliminating single-source dependencies that often plague complex intermediate synthesis. The robustness of the reaction conditions, which tolerate a wide range of functional groups and do not require cryogenic temperatures or ultra-high pressures, simplifies the engineering requirements for production facilities. This ease of operation translates to faster turnaround times and the ability to ramp up production volume rapidly in response to market demand, effectively reducing lead time for high-purity pharmaceutical intermediates.
Scalability and environmental compliance are also markedly improved with this technology. The use of a solid CO surrogate (TFBen) eliminates the need for handling toxic carbon monoxide gas, significantly enhancing workplace safety and reducing the regulatory burden associated with hazardous gas storage and usage. The high atom economy and clean reaction profile minimize the generation of hazardous waste, aligning with green chemistry principles and reducing disposal costs. The process has been demonstrated to be scalable, providing a clear path from gram-scale discovery to multi-ton commercial production, thereby securing the supply continuity essential for cost reduction in pharmaceutical intermediate manufacturing and supporting the uninterrupted delivery of critical drug substances.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method, derived directly from the patent specifications and experimental data. These insights are intended to clarify the operational feasibility and strategic value of adopting this technology for your specific production needs. Understanding these details will help stakeholders make informed decisions about integrating this route into their existing manufacturing portfolios.
Q: What are the primary advantages of this novel synthesis method over conventional cyclization routes?
A: Unlike traditional methods that rely on harsh conditions, expensive pre-activated substrates like isatoic anhydride, or unstable trifluoroacetamides, this novel approach utilizes cheap and readily available trifluoroethylimidoyl chloride. It operates under milder conditions with significantly higher reaction efficiency and broader substrate compatibility, allowing for the synthesis of diverse derivatives without complex pre-functionalization steps.
Q: Can this methodology be applied to the synthesis of complex bioactive drug molecules?
A: Yes, the patent explicitly demonstrates the successful application of this method in the high-yield total synthesis of Rutaecarpine, a potent natural product with significant pharmacological activity. The process achieved an overall yield of 77% over three steps, proving its robustness for constructing complex fused-ring heterocyclic systems found in active pharmaceutical ingredients.
Q: What is the scope of substrates compatible with this palladium-catalyzed reaction?
A: The reaction exhibits excellent functional group tolerance, accommodating a wide range of amines including alkyl, cycloalkyl, benzyl, and substituted aryl amines. Furthermore, the aromatic ring of the imidoyl chloride can bear various substituents such as halogens, methyl groups, or trifluoromethyl groups at ortho, meta, or para positions, making it highly versatile for medicinal chemistry optimization.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust and scalable synthetic routes in the fast-paced pharmaceutical industry. Our team of expert process chemists has thoroughly evaluated the technology described in patent CN113045503B and is fully equipped to leverage this advanced palladium-catalyzed carbonylation method for your projects. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. Our state-of-the-art facilities are designed to handle complex organometallic reactions with the highest standards of safety and quality control.
We are committed to delivering stringent purity specifications through our rigorous QC labs, which utilize advanced analytical techniques to verify the identity and purity of every batch. Whether you require custom synthesis of novel quinazolinone derivatives or the commercial supply of established intermediates like those used in Rutaecarpine synthesis, we offer tailored solutions to meet your exact requirements. We invite you to contact our technical procurement team today to request a Customized Cost-Saving Analysis. Let us provide you with specific COA data and route feasibility assessments to demonstrate how we can optimize your supply chain and drive down costs while maintaining the highest quality standards.