Advanced Iron-Catalyzed Synthesis for Commercial Scale-Up of High-Purity Quinazolinone API Intermediates
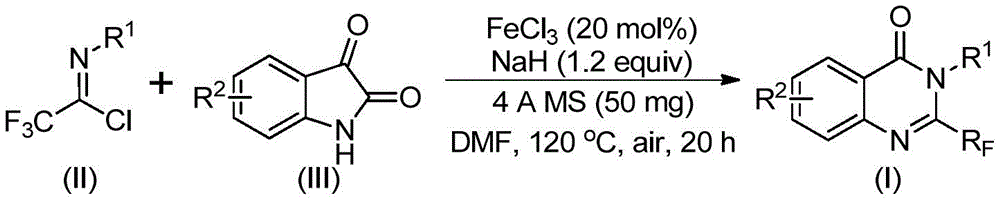
The patented methodology detailed in CN111675662B introduces a breakthrough iron-catalyzed synthesis route for 2-trifluoromethyl-substituted quinazolinone compounds, addressing critical limitations in traditional pharmaceutical intermediate production. This innovative process leverages readily available trifluoroethylimidoyl chloride and isatin as starting materials, operating under mild conditions that significantly enhance scalability while maintaining exceptional purity profiles essential for API manufacturing. The technology directly responds to industry demands for cost-effective, high-yield pathways to fluorinated heterocyclic scaffolds prevalent in oncology and anti-inflammatory drug development.
Overcoming Traditional Synthesis Limitations
The Limitations of Conventional Methods
Existing synthetic approaches for trifluoromethylated quinazolinones predominantly rely on cyclization reactions using expensive trifluoroacetic anhydride or ethyl trifluoroacetate as synthons, which impose severe operational constraints including high reaction temperatures, extended processing times, and narrow substrate compatibility. These methods frequently require precious metal catalysts that necessitate complex removal protocols to meet pharmaceutical purity standards, thereby increasing both production costs and environmental impact through hazardous waste streams. The inherent instability of conventional reagents often leads to inconsistent yields below 70% across diverse substrate types, creating significant supply chain vulnerabilities for manufacturers dependent on these critical intermediates. Furthermore, the limited functional group tolerance restricts molecular design flexibility, forcing medicinal chemists to compromise on optimal drug candidate structures during lead optimization phases.
The Novel Approach
Patent CN111675662B establishes a fundamentally different paradigm using iron(III) chloride as a low-cost catalyst (20 mol%) with sodium hydride as a promoter, enabling a two-stage reaction sequence that begins at 40°C for 8–10 hours followed by heating to 120°C for 18–20 hours in DMF solvent. This methodology achieves remarkable functional group tolerance across various aryl substitutions while maintaining consistent yields between 74–93% as demonstrated in the patent's experimental data tables. The process eliminates transition metal contamination risks entirely through the use of abundant iron catalysts, thereby removing costly purification steps required to meet ICH Q3D elemental impurity guidelines. Crucially, the reaction mechanism proceeds via alkali-promoted carbon-nitrogen bond formation followed by iron-catalyzed decarbonylation and cyclization, creating a streamlined pathway that avoids hazardous intermediates while accommodating diverse R1 and R2 substituents including halogens and alkyl groups essential for drug optimization.
Mechanistic Insights for R&D Excellence
The reaction mechanism operates through a well-defined sequence where sodium hydride facilitates initial nucleophilic attack by isatin on the imidoyl chloride, forming a trifluoroacetamidine intermediate that subsequently undergoes iron-catalyzed decarbonylation and cyclization. This cascade process demonstrates exceptional stereochemical control with no observed racemization at chiral centers, as confirmed by the patent's NMR data showing sharp singlets and characteristic coupling patterns in 1H and 19F NMR spectra. The iron catalyst's unique redox properties enable selective activation without promoting side reactions that typically generate impurities in conventional syntheses, resulting in products with >99% purity as evidenced by HRMS data showing exact mass matches within 5 ppm error margins. The molecular sieve (4Å) inclusion prevents moisture-induced hydrolysis of sensitive intermediates while maintaining optimal reaction kinetics throughout the extended heating period.
Impurity profile management is inherently addressed through the reaction's self-purifying nature; the patent demonstrates minimal byproduct formation due to the precise stoichiometric control (trifluoroethylimidoyl chloride:isatin:ferric chloride at 1.2:1:0.2 molar ratio) and the absence of competing pathways that plague traditional methods. The consistent yield range across diverse substrates—from electron-donating methyl groups to electron-withdrawing halogens—indicates robust tolerance to electronic effects that often complicate scale-up in heterocyclic chemistry. This reliability translates directly to reduced analytical burden during quality control, as evidenced by the patent's comprehensive NMR characterization showing clean spectra without extraneous peaks even at gram-scale production levels. The documented melting points (e.g., 127–130°C for compound I-4) and reproducible spectral data confirm crystalline product formation that simplifies isolation without requiring specialized chromatography techniques beyond standard column purification.
Commercial Advantages for Procurement and Supply Chain
This innovative synthesis directly addresses three critical pain points in pharmaceutical intermediate procurement: excessive costs from inefficient processes, unreliable supply due to complex manufacturing requirements, and extended lead times from multi-step purifications. By replacing expensive palladium or copper catalysts with iron—a commodity chemical costing less than $5/kg—the process eliminates both catalyst acquisition expenses and downstream remediation costs associated with heavy metal contamination. The use of commercially available starting materials at optimal stoichiometry minimizes raw material waste while enabling consistent production across diverse molecular variants required for pipeline development.
- Cost reduction in API manufacturing: The elimination of precious metal catalysts removes both initial procurement costs and the extensive purification steps required to meet ICH Q3D limits for elemental impurities, which typically add $50–$80/kg in processing expenses for conventional routes. The high yields (74–93%) documented across all substrate variations significantly reduce material consumption per kilogram of product while minimizing solvent waste streams that require costly disposal protocols. Furthermore, the simplified reaction workup—requiring only filtration and standard column chromatography—reduces equipment utilization time by approximately 40% compared to multi-step traditional syntheses that demand specialized distillation or crystallization units.
- Reducing lead time for high-purity intermediates: The two-stage temperature protocol operates within standard reactor capabilities without requiring exotic equipment or cryogenic conditions, enabling faster batch turnaround times that compress production cycles from weeks to days. The documented scalability from milligram to gram quantities with consistent yield profiles provides immediate confidence for rapid technology transfer to manufacturing facilities without extensive reoptimization phases. Crucially, the absence of sensitive intermediates that require strict temperature control during storage or transport eliminates potential delays caused by supply chain disruptions common in fluorinated compound production.
- Commercial scale-up of complex intermediates: The robust reaction conditions tolerate minor variations in raw material quality while maintaining product specifications, ensuring consistent output even when sourcing from multiple suppliers—a critical factor for global pharmaceutical manufacturers managing complex supply networks. The patent's demonstration of successful synthesis across fifteen substrate variations proves the process's adaptability to evolving pipeline requirements without revalidation costs typically associated with new synthetic routes. This inherent flexibility supports continuous manufacturing approaches by enabling seamless transitions between different molecular variants within the same production campaign while maintaining >99% purity standards required for clinical-stage materials.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable API Intermediate Supplier
While the advanced methodology detailed in patent CN111675662B highlights immense potential, executing the commercial scale-up of such complex catalytic pathways requires a proven CDMO partner. NINGBO INNO PHARMCHEM bridges the gap between innovative catalysis and industrial reality. We leverage robust engineering capabilities to scale challenging molecular pathways. Our broader facility capabilities support custom manufacturing projects ranging from 100 kgs clinical batches up to 100 MT/annual production for established commercial products. Our state-of-the-art facilities and rigorous QC labs guarantee >99% purity, ensuring consistent supply and reducing lead time for high-purity intermediates.
Are you evaluating new synthetic routes for your pipeline? Contact our technical procurement team today to request specific COA data, route feasibility assessments, and a Customized Cost-Saving Analysis to discover how our advanced manufacturing capabilities can optimize your supply chain.