Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles: A Breakthrough for Commercial API Manufacturing
Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles: A Breakthrough for Commercial API Manufacturing
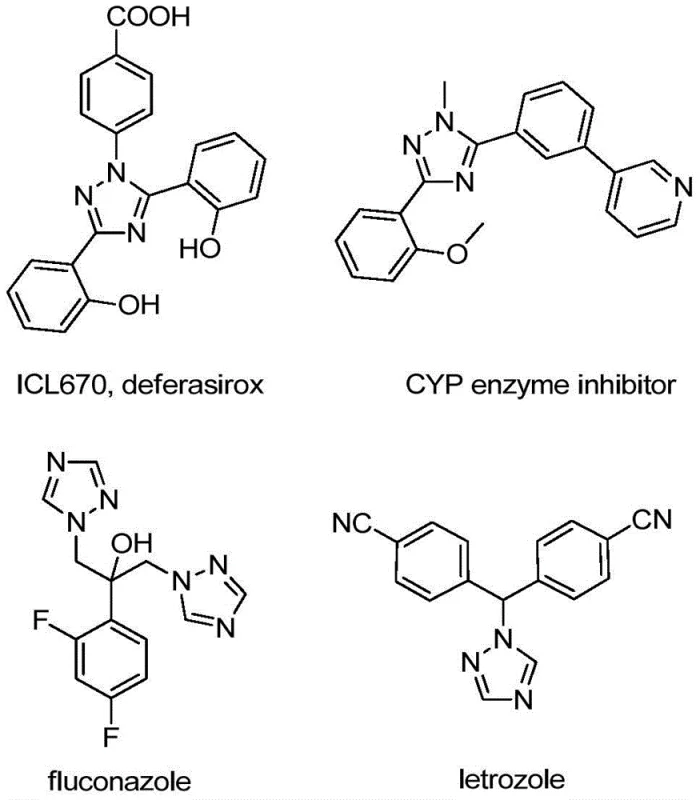
The pharmaceutical and agrochemical industries are constantly seeking robust, scalable, and cost-effective methodologies for constructing nitrogen-containing heterocycles, particularly those bearing trifluoromethyl groups which enhance metabolic stability and lipophilicity. Patent CN110467579B discloses a highly efficient preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds, addressing critical bottlenecks in traditional synthetic routes. This technology leverages a non-metallic iodine-promoted cyclization strategy that operates under mild conditions, eliminating the need for expensive transition metal catalysts or rigorous anhydrous environments. For R&D directors and procurement managers alike, this represents a significant opportunity to streamline the supply chain for high-value intermediates used in drugs like fluconazole and letrozole, ensuring both purity and economic viability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethylated nitrogen heterocycles has relied heavily on two primary strategies, both of which present substantial challenges for large-scale manufacturing. The first approach involves the direct trifluoromethylation of pre-synthesized heterocyclic cores, which often necessitates the use of specialized and costly trifluoromethylating reagents that can be hazardous to handle and difficult to source in bulk quantities. The second mainstream method utilizes trifluoromethyl synthons, such as trifluorodiazoethane or trifluoroethylimide halides, but these reactions frequently require harsh conditions, sensitive catalysts, or complex protection-deprotection sequences. Furthermore, many conventional protocols demand strictly anhydrous and anaerobic conditions to prevent side reactions, imposing severe infrastructure costs on production facilities and complicating the operational workflow for process chemists aiming for commercial scale-up.
The Novel Approach
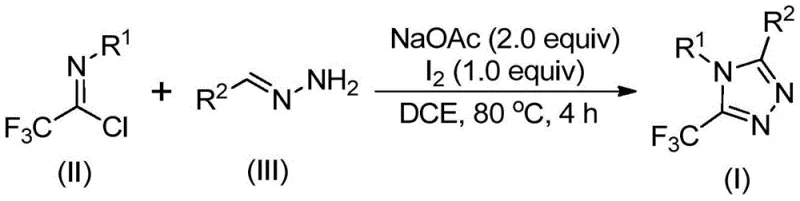
In stark contrast, the methodology outlined in patent CN110467579B introduces a streamlined, one-pot synthesis that utilizes inexpensive and readily available starting materials: hydrazones and trifluoroethylimidoyl chloride. By employing elemental iodine as a promoter rather than a stoichiometric oxidant or heavy metal catalyst, this novel route achieves high conversion rates under relatively benign thermal conditions. The process tolerates a wide range of functional groups on both the hydrazone and the imidoyl chloride substrates, allowing for the facile synthesis of diverse 4,5-disubstituted 1,2,4-triazole derivatives. This flexibility not only accelerates library generation for drug discovery but also simplifies the regulatory pathway for commercial production by minimizing the risk of toxic metal contamination in the final active pharmaceutical ingredient.
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of base-promoted condensation and oxidative aromatization, driven by the unique reactivity of elemental iodine in the presence of sodium acetate. Initially, the reaction likely proceeds through a base-mediated intermolecular carbon-nitrogen bond formation between the hydrazone and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine intermediate. This species then undergoes isomerization to align the reactive centers for cyclization. The critical step involves the oxidative iodination promoted by the base and elemental iodine, which generates an iodinated intermediate that serves as a potent electrophile. This sets the stage for an intramolecular electrophilic substitution reaction, effectively closing the triazole ring. Finally, the system undergoes spontaneous aromatization to yield the stable 5-trifluoromethyl-1,2,4-triazole core, releasing hydrogen iodide which is neutralized by the acetate base.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based trifluoromethylation methods. Because the cyclization is driven by polar mechanisms and electrophilic aromatic substitution rather than free-radical chains, the formation of random polymeric byproducts or regio-isomeric impurities is significantly suppressed. The use of sodium acetate as a mild buffer helps maintain a consistent pH environment, preventing the hydrolysis of the sensitive imidoyl chloride starting material while facilitating the deprotonation steps required for ring closure. Consequently, the crude reaction mixture typically exhibits a cleaner profile, reducing the burden on downstream purification units such as column chromatography or recrystallization, which is a critical factor for maintaining high overall yields in multi-kilogram batches.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Efficiently
The practical execution of this synthesis is designed for operational simplicity, making it accessible for both laboratory-scale optimization and pilot-plant production. The protocol involves charging a reaction vessel with sodium acetate, trifluoroethylimidoyl chloride, and the corresponding hydrazone in a suitable organic solvent like 1,2-dichloroethane (DCE). The mixture is heated to 80°C for an initial period to allow the condensation to proceed, after which elemental iodine is introduced to drive the oxidative cyclization to completion. Detailed standardized operating procedures regarding exact stoichiometry, safety handling of iodine, and specific workup protocols are essential for reproducibility.
- Combine sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane (DCE) within a reaction vessel.
- Heat the reaction mixture to 80°C and maintain stirring for approximately 3 hours to facilitate the initial condensation and cyclization steps.
- Add elemental iodine to the system and continue heating for an additional 1 hour to promote oxidative aromatization, followed by standard filtration and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis route offers transformative benefits that extend far beyond simple yield improvements. The elimination of precious metal catalysts such as palladium or copper removes a major cost driver and supply chain vulnerability, as these metals are subject to significant price volatility and geopolitical sourcing risks. Furthermore, the ability to run the reaction without stringent anhydrous or oxygen-free conditions drastically reduces the capital expenditure required for specialized reactor setups and inert gas purging systems, allowing existing general-purpose manufacturing lines to be utilized effectively. This operational flexibility translates directly into reduced lead times and enhanced supply continuity for critical pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The economic model of this process is underpinned by the use of commodity-grade starting materials. Hydrazones can be synthesized in situ or purchased cheaply from aldehydes and hydrazine hydrate, while trifluoroethylimidoyl chloride is a stable, commercially available building block. By replacing expensive trifluoromethylating reagents and noble metal catalysts with inexpensive sodium acetate and elemental iodine, the direct material cost per kilogram of product is significantly lowered. Additionally, the simplified workup procedure, which avoids complex metal scavenging steps, reduces solvent consumption and waste disposal costs, contributing to a leaner and more profitable manufacturing process.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the broad availability of the requisite reagents. Unlike specialized fluorinating agents that may have limited suppliers and long lead times, the components for this reaction are standard chemicals found in the inventory of most fine chemical distributors. The robustness of the reaction conditions means that production is less susceptible to delays caused by environmental factors or minor deviations in utility quality. This reliability ensures that downstream API manufacturers can maintain consistent production schedules, mitigating the risk of stockouts for high-demand medications that rely on this triazole scaffold.
- Scalability and Environmental Compliance: The scalability of this method is evidenced by its successful demonstration at the gram level with high efficiency, indicating a clear path to kilogram and ton-scale production. The absence of heavy metals simplifies the environmental compliance landscape, as there is no need for extensive wastewater treatment to remove toxic metal residues. The use of elemental iodine, which can potentially be recovered or managed more easily than organometallic waste, aligns with modern green chemistry principles. This makes the process not only easier to scale but also more sustainable, helping companies meet increasingly stringent corporate social responsibility and environmental, social, and governance (ESG) targets.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a clear understanding of the process capabilities and limitations for potential adopters.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN110467579B utilizes elemental iodine as a promoter instead of expensive heavy metal catalysts, significantly simplifying downstream purification and reducing heavy metal residue concerns.
Q: What are the optimal reaction conditions for this triazole synthesis?
A: The optimal conditions involve heating the reactants in dichloroethane (DCE) at 80°C for 3 hours, followed by the addition of iodine and further reaction for 1 hour, achieving yields up to 97% for specific substrates.
Q: Is strict anhydrous or anaerobic conditioning necessary for this process?
A: One of the key advantages of this protocol is that it does not require stringent anhydrous or oxygen-free conditions, making it highly suitable for large-scale industrial operations where such controls add significant cost.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient heterocycle synthesis in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to full-scale manufacturing is seamless and compliant. We are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole intermediate meets the exacting standards required for global pharmaceutical registration.
We invite you to leverage our technical expertise to optimize your supply chain for these critical building blocks. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our implementation of this iodine-promoted technology can drive value and reliability for your organization.