Advanced Olmesartan Medoxomil Manufacturing Technology Enhancing Purity and Commercial Scalability for Global Pharma
The pharmaceutical industry continuously seeks robust synthetic pathways for Angiotensin II Receptor Blockers (ARBs), and the technology disclosed in Chinese Patent CN1271068C represents a significant advancement in the manufacturing of Olmesartan Medoxomil. This specific intellectual property outlines a novel preparation method that fundamentally restructures the synthetic sequence to enhance overall efficiency and product quality. By strategically modifying the order of functional group transformations, the process mitigates the formation of complex impurity profiles that often plague traditional synthesis routes. The core innovation lies in the ring-opening strategy of a furoimidazolone intermediate, which serves as a pivotal precursor for the final active pharmaceutical ingredient. This approach not only streamlines the reaction sequence but also aligns with the rigorous demands of modern Good Manufacturing Practices (GMP) for high-purity olmesartan medoxomil. As a reliable olmesartan medoxomil supplier, understanding these underlying chemical mechanisms is crucial for ensuring supply chain resilience and product consistency.
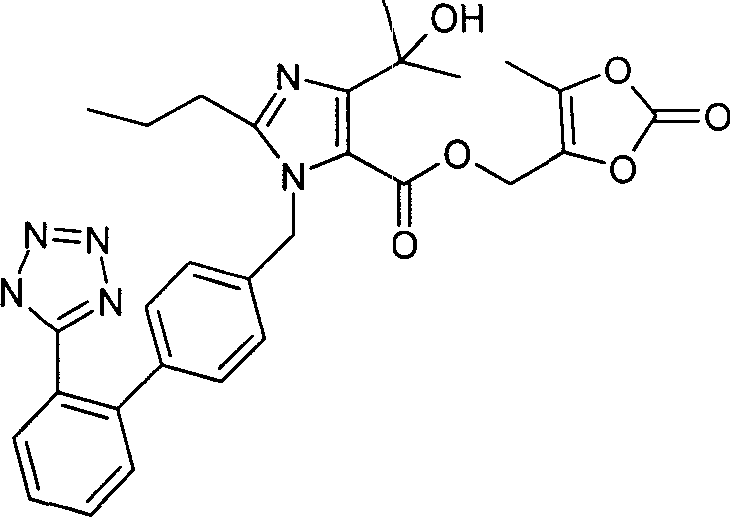
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Olmesartan Medoxomil has been challenged by the inherent reactivity of the hydroxyl group located at the 4-position of the imidazole ring. In many prior art methods, such as those described in Japanese Patent JP (31) 27098 and European Patent EP503785, the imidazole and biphenyl moieties are connected early in the synthetic sequence. This early coupling creates a chemical environment where the hydroxyl group is prone to unwanted etherification with the biphenyl component. Such side reactions lead to a complex mixture of by-products that are structurally similar to the target molecule, making subsequent purification steps arduous and costly. The low selectivity inherent in these conventional routes often results in diminished overall yields, requiring extensive chromatographic separation or recrystallization processes that increase waste generation and production time. Furthermore, the harsh conditions sometimes required to drive these reactions to completion can compromise the stability of the sensitive tetrazole ring, leading to further degradation and impurity formation.
The Novel Approach
In contrast, the method disclosed in CN1271068C introduces a strategic reversal of the synthetic logic to overcome these selectivity issues. By starting with a protected furoimidazolone derivative, the process effectively masks the reactive centers until the appropriate stage of the synthesis. The key breakthrough involves the ring-opening of this cyclic precursor to generate the carboxylic acid intermediate only after the core imidazole structure is securely established. This sequence ensures that the hydroxyl group is not exposed to etherification conditions prematurely, thereby drastically reducing the formation of side products. The result is a cleaner reaction profile that simplifies downstream processing and enhances the isolation of the target compound. This novel approach demonstrates a more rational design of the synthetic pathway, prioritizing chemoselectivity and operational simplicity, which are critical factors for achieving cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Ring-Opening Hydrolysis and Esterification
The first critical transformation in this novel pathway involves the hydrolysis of the furoimidazolone ring, a step that converts the cyclic Compound (A) into the open-chain carboxylic acid Compound (B). This reaction is typically facilitated by the use of alkali metal hydroxides or carbonates in a solvent system comprising alcohols, ethers, or amides. The mechanism proceeds through a nucleophilic attack on the carbonyl carbon of the lactone ring, leading to ring cleavage and the formation of the carboxylate salt. The beauty of this step lies in its robustness; the patent indicates that the reaction temperature can vary widely from -20°C to 120°C without significantly impacting the outcome, suggesting a low activation energy barrier and high tolerance to process variations. This flexibility is invaluable for commercial scale-up of complex ARB intermediates, as it allows manufacturers to optimize thermal inputs based on available equipment and energy constraints.
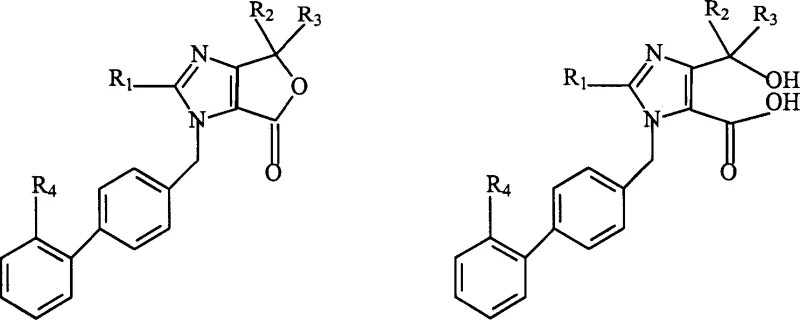
Following the generation of the carboxylic acid intermediate, the synthesis proceeds to the esterification step, which installs the medoxomil moiety essential for the drug's bioavailability. This transformation involves the reaction of the acid intermediate with 4-bromo(or chloro)methyl-5-methyl-2-oxo-1,3-dioxolene in the presence of a base catalyst. The reaction conditions are notably mild, typically operating between 0°C and 120°C, with a preferred range of 20°C to 80°C, ensuring that the sensitive tetrazole protecting group remains intact. The final step involves the acidic removal of the trityl protecting group to reveal the active tetrazole functionality. This deprotection is conducted under controlled acidic conditions using organic or inorganic acids, completing the synthesis of Olmesartan Medoxomil. The entire sequence is designed to minimize exposure of the molecule to harsh environments, preserving the integrity of the final product.
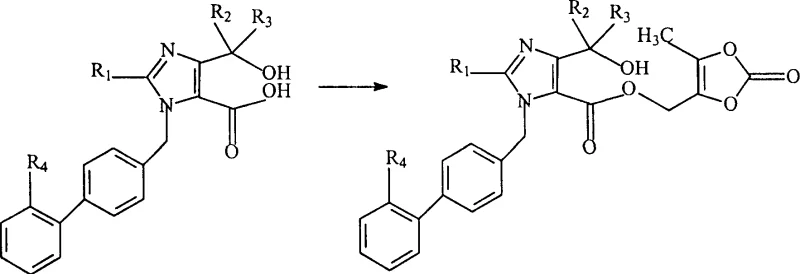
How to Synthesize Olmesartan Medoxomil Efficiently
Implementing this synthesis route requires careful attention to solvent selection and stoichiometry to maximize the benefits of the novel pathway. The process begins with the preparation of the furoimidazolone precursor, which can be sourced or synthesized according to related patent literature. The subsequent hydrolysis and esterification steps must be monitored to ensure complete conversion while avoiding over-reaction that could lead to degradation. Detailed standard operating procedures are essential for maintaining batch-to-batch consistency, particularly during the workup and purification phases where the removal of inorganic salts and solvent residues is critical. For a comprehensive guide on the specific operational parameters and safety considerations, please refer to the standardized synthesis steps provided below.
- Hydrolyze the furoimidazolone precursor (Compound A) using alkali or acid in a solvent system to obtain the open-ring carboxylic acid intermediate (Compound B).
- React the resulting carboxylic acid salt or free acid with 4-bromo(or chloro)methyl-5-methyl-2-oxo-1,3-dioxolene under basic catalysis to form the ester linkage.
- Remove the trityl protecting group from the tetrazole moiety using an acid catalyst to yield the final Olmesartan Medoxomil product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented synthesis method offers substantial benefits for procurement and supply chain management teams seeking to optimize their sourcing strategies. The simplified reaction sequence reduces the number of unit operations required, which directly translates to lower operational expenditures and reduced consumption of raw materials. By minimizing the formation of difficult-to-remove by-products, the process decreases the reliance on expensive purification media and solvents, contributing to a more sustainable and cost-effective manufacturing model. Additionally, the mild reaction conditions reduce the energy load on production facilities, further enhancing the economic viability of the process. These factors collectively support the goal of reducing lead time for high-purity antihypertensive agents by streamlining the production cycle.
- Cost Reduction in Manufacturing: The elimination of complex purification steps required to remove etherification by-products significantly lowers the cost of goods sold. By avoiding the use of expensive transition metal catalysts often found in cross-coupling reactions, the process relies on readily available inorganic bases and acids, which are cost-efficient and easy to source. The high selectivity of the reaction ensures that raw material utilization is maximized, reducing waste disposal costs and improving overall atom economy. This logical deduction of cost savings is based on the mechanistic simplicity and operational efficiency inherent in the patented route.
- Enhanced Supply Chain Reliability: The reagents required for this synthesis, such as alkali metal hydroxides, carbonates, and common organic solvents like acetone and DMF, are commodity chemicals with stable global supply chains. This reduces the risk of production delays caused by the scarcity of specialized or exotic reagents. Furthermore, the robustness of the reaction conditions allows for manufacturing in diverse geographic locations without the need for highly specialized equipment, enhancing supply chain resilience. The ability to source raw materials easily ensures continuous production capability and mitigates the risk of supply disruptions.
- Scalability and Environmental Compliance: The process is explicitly designed for industrial production, with reaction parameters that are easily scalable from laboratory to commercial plant sizes. The reduction in side reactions means less chemical waste is generated, simplifying effluent treatment and ensuring compliance with increasingly stringent environmental regulations. The use of standard solvents and reagents facilitates solvent recovery and recycling programs, further minimizing the environmental footprint. This alignment with green chemistry principles makes the process attractive for manufacturers aiming to meet sustainability goals while maintaining high production volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Olmesartan Medoxomil synthesis technology. These answers are derived directly from the technical specifications and beneficial effects described in the patent documentation. They are intended to provide clarity on the process capabilities and its advantages over traditional methods. For further technical details or specific data regarding impurity profiles, please consult the full patent text or contact our technical team.
Q: How does this novel method improve selectivity compared to conventional routes?
A: Conventional methods often link the imidazole and biphenyl parts early, causing etherification side reactions at the 4-position hydroxyl group. This patent's method delays this connection, significantly reducing by-products and simplifying purification.
Q: What are the typical reaction conditions for the hydrolysis step?
A: The hydrolysis of the furoimidazolone ring can be conducted over a broad temperature range from -20°C to 120°C, with a preferred range of 0°C to 100°C, allowing for flexible process control.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method features simple operation, mild reaction conditions, and high yield, making it highly suitable for industrial implementation and commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Olmesartan Medoxomil Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and reliable synthesis routes for key pharmaceutical intermediates like Olmesartan Medoxomil. Our team of expert chemists has extensively evaluated the technology disclosed in CN1271068C and confirmed its potential for robust commercial manufacturing. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory scale to industrial volume is seamless and efficient. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch meets the highest quality standards required by global regulatory authorities.
We invite procurement leaders and technical directors to collaborate with us to leverage this advanced synthesis technology for their supply chains. By partnering with us, you can request a Customized Cost-Saving Analysis to quantify the economic benefits of switching to this more efficient route. Our technical procurement team is ready to provide specific COA data and comprehensive route feasibility assessments tailored to your specific production requirements. Let us help you optimize your manufacturing strategy and secure a stable supply of high-quality Olmesartan Medoxomil.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →