Scalable Manufacturing of Etoposide Phosphate: A Technical Breakdown for Global Supply Chains
Scalable Manufacturing of Etoposide Phosphate: A Technical Breakdown for Global Supply Chains
The development of water-soluble prodrugs for poorly soluble anticancer agents remains a critical challenge in oncology formulation, particularly for the topoisomerase II inhibitor etoposide. Patent CN1033589C, filed in late 1996, introduces a robust preparation method for etoposide phosphate that addresses significant scalability issues found in earlier art. This technical insight report analyzes the proprietary synthetic route which utilizes a haloacetyl protection strategy to achieve high regioselectivity and yield. By shifting away from complex chromatographic purifications and lyophilization steps, this methodology offers a compelling value proposition for reliable pharmaceutical intermediate suppliers aiming to secure long-term contracts with generic API manufacturers. The following analysis details the chemical mechanisms, process advantages, and supply chain implications of adopting this specific synthetic pathway for commercial production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those disclosed in US Patent 4,904,768, typically involve the direct reaction of etoposide with phosphoryl chloride or alkyl chlorophosphates. While conceptually straightforward, this approach suffers from severe selectivity issues; the phosphorus compound reacts not only with the target 4'-hydroxyl group but also continuously with the hydroxyl groups on the sugar moiety. This lack of chemoselectivity generates a complex mixture of byproducts, necessitating rigorous purification techniques like column chromatography to isolate the target etoposide phosphate. Furthermore, alternative methods described in Japanese publication No. 192793/1988 utilize acetate protection groups which require removal using zinc and acetic acid. This reductive deprotection step creates significant downstream processing burdens, specifically requiring lyophilization to separate the final product, a technique that is energy-intensive and notoriously difficult to implement efficiently in large-scale industrial reactors.
The Novel Approach
The methodology outlined in CN1033589C circumvents these bottlenecks by employing a haloacetyl group (such as dichloroacetyl) to protect the sugar hydroxyls prior to phosphorylation. This strategic modification ensures that the phosphorylation reaction occurs exclusively at the 4'-position of the aglycone, effectively suppressing side reactions on the glycosidic ring. Crucially, the removal of the haloacetyl protecting group is achieved through solvolysis in the presence of an amine, such as triethylamine, under mild conditions. This eliminates the need for heavy metal reductants like zinc and avoids the thermal stress of lyophilization. The result is a process where the final product can be isolated via simple crystallization and filtration, drastically reducing solvent consumption and processing time while maintaining a high purity profile suitable for regulatory submission.
Mechanistic Insights into Haloacetyl-Mediated Regioselective Phosphorylation
The core innovation of this synthesis lies in the orthogonal stability of the haloacetyl protecting group. In the initial stages, the 2,3-dihydroxyl groups of the glucose moiety in 4'-demethylepipodophyllotoxin are esterified with a halogenated acyl chloride, such as dichloroacetyl chloride. This steric and electronic modification renders the sugar hydroxyls inert to the subsequent phosphorylation conditions. When the protected intermediate is treated with phosphoryl chloride (POCl3) in the presence of a tertiary amine base at cryogenic temperatures ranging from -15°C to -5°C, the electrophilic phosphorus species attacks solely the more nucleophilic 4'-phenolic hydroxyl. This step forms a stable dichlorophosphate intermediate, which is then carefully hydrolyzed to the monophosphate. The final deprotection step exploits the lability of the alpha-halo ester bond; treatment with a primary, secondary, or tertiary amine in an alcoholic solvent facilitates a nucleophilic acyl substitution that cleaves the protecting group without affecting the newly formed phosphate ester bond.
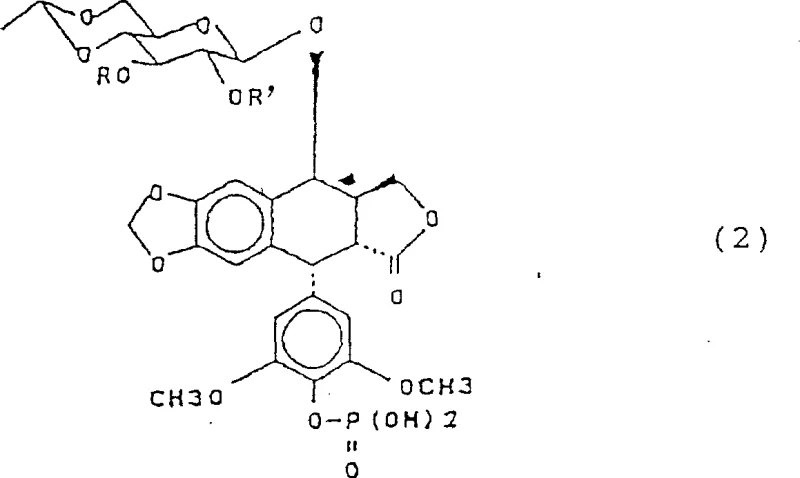
As illustrated in the structural representation of the key intermediate (Formula 2), the presence of the -COCHmX(3-m) groups on the sugar ring is the defining feature that enables this high-yield transformation. The electron-withdrawing nature of the halogen atoms increases the electrophilicity of the carbonyl carbon, making it susceptible to aminolysis by reagents like triethylamine or diethylamine at temperatures as low as 0°C to 30°C. This mechanistic pathway prevents the epimerization of the sensitive C-4 position on the lignan backbone, a common degradation pathway in basic conditions. By controlling the pH during the workup—specifically adjusting to pH 1.0-2.5 to extract the product into the organic phase—the process ensures that the final etoposide phosphate is obtained with minimal isomeric impurities, as evidenced by the low isomer content (e.g., 0.6%) reported in the experimental embodiments.
How to Synthesize Etoposide Phosphate Efficiently
The synthesis of this high-value oncology intermediate requires precise control over reaction stoichiometry and temperature gradients to maximize the overall yield, which the patent reports can reach up to 68.4% over three steps. The process begins with the coupling of protected podophyllotoxin derivatives followed by the critical phosphorylation and deprotection sequence described above. Operators must maintain strict anhydrous conditions during the phosphorylation step to prevent premature hydrolysis of the phosphoryl chloride, while the final deprotection requires careful monitoring of amine equivalents (typically 1-3 equivalents) to ensure complete removal of the haloacetyl groups without degrading the phosphate ester. For a detailed breakdown of the specific reagent grades, solvent swaps, and crystallization parameters required to replicate this high-purity output, please refer to the standardized operating procedure below.
- Protect the sugar moiety hydroxyl groups of 4'-demethylepipodophyllotoxin with a haloacetyl group (e.g., dichloroacetyl) to prevent side reactions during phosphorylation.
- React the protected intermediate with phosphoryl chloride (POCl3) in the presence of a tertiary amine base at low temperatures (-15°C to -5°C) to introduce the phosphate group at the 4' position.
- Hydrolyze the dichlorophosphate intermediate and subsequently remove the haloacetyl protecting groups using an amine (such as triethylamine) in alcohol solvent to yield the final etoposide phosphate product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition from chromatography-dependent processes to crystallization-based isolation represents a fundamental shift in cost structure and risk management. Traditional methods relying on column chromatography are inherently batch-limited, solvent-heavy, and prone to variability, which translates to unpredictable lead times and high unit costs. In contrast, the haloacetyl protection route described in CN1033589C enables a continuous or large-batch workflow where purification is driven by physical phase changes (crystallization) rather than adsorption dynamics. This shift significantly reduces the consumption of high-purity silica gel and elution solvents, directly lowering the variable cost of goods sold (COGS). Furthermore, the elimination of lyophilization removes a major bottleneck in drying capacity, allowing manufacturing facilities to increase throughput without capital expenditure on specialized freeze-drying equipment.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the removal of expensive purification steps. By avoiding column chromatography, the manufacturer eliminates the cost of stationary phases and the extensive solvent recovery systems required to handle large elution volumes. Additionally, the use of inexpensive amines like triethylamine for deprotection, rather than costly catalytic hydrogenation or metal reductions, further optimizes the raw material spend. The ability to isolate the product via precipitation with non-polar solvents like n-hexane allows for efficient solvent recycling, creating a closed-loop system that minimizes waste disposal fees and enhances the overall margin profile for high-purity pharmaceutical intermediate production.
- Enhanced Supply Chain Reliability: Supply continuity is often compromised by complex purification steps that have low tolerance for feedstock variability. This novel method demonstrates robustness against minor fluctuations in starting material quality because the crystallization step acts as a powerful purification engine, rejecting impurities into the mother liquor. The reliance on commodity chemicals such as phosphoryl chloride, triethylamine, and common alcohols ensures that the supply chain is not vulnerable to shortages of exotic reagents. This stability allows suppliers to offer more reliable delivery schedules and maintain safety stock levels with greater confidence, mitigating the risk of production stoppages that could disrupt the downstream API synthesis for global oncology markets.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, the replacement of zinc/acetic acid reduction with amine solvolysis drastically reduces the heavy metal load in the wastewater stream. This simplifies effluent treatment and ensures compliance with increasingly stringent environmental regulations regarding heavy metal discharge. The process is inherently scalable from kilogram to multi-ton quantities because the unit operations involved—cooling, filtration, and crystallization—are standard in modern cGMP facilities. The high yields reported (up to 97% for intermediate steps) mean that less raw material is wasted per kilogram of output, aligning with green chemistry principles and reducing the carbon footprint associated with the commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis route in a commercial setting. These answers are derived directly from the experimental data and claims within the patent documentation, focusing on the practical aspects of yield optimization, impurity control, and process safety. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer or contract manufacturing agreements.
Q: Why is haloacetyl protection superior to acetate protection for etoposide phosphate synthesis?
A: Haloacetyl groups allow for mild deprotection using amines under neutral or slightly acidic conditions, avoiding the harsh zinc/acetic acid reduction required for acetate groups. This eliminates the need for lyophilization and simplifies purification, making it ideal for large-scale manufacturing.
Q: How does this method improve impurity profiles compared to direct phosphorylation?
A: Direct phosphorylation often leads to unwanted phosphorylation of sugar hydroxyls, creating difficult-to-remove byproducts that require column chromatography. The haloacetyl protection strategy blocks these sugar hydroxyls, ensuring regioselectivity at the 4'-position and allowing purification via simple crystallization.
Q: What are the critical temperature controls in the phosphorylation step?
A: The reaction with phosphoryl chloride must be conducted at low temperatures, typically between -15°C and -5°C, to control exothermicity and prevent degradation of the sensitive podophyllotoxin backbone, ensuring high yields of the dichlorophosphate intermediate.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Etoposide Phosphate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of oncology drugs depends on a supply chain that balances technical precision with economic efficiency. Our R&D team has extensively analyzed the protective group strategies detailed in CN1033589C and validated their applicability for large-scale production. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to plant-scale manufacturing is seamless. Our facility is equipped with state-of-the-art cryogenic reactors capable of maintaining the strict -15°C conditions required for the phosphorylation step, alongside rigorous QC labs that enforce stringent purity specifications to meet the exacting standards of global regulatory bodies.
We invite procurement leaders and technical directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By leveraging our optimized version of this haloacetyl protection route, we can help you reduce lead time for high-purity pharmaceutical intermediates while securing a stable supply of this critical cancer therapy component. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing expertise can become a strategic asset in your supply chain.