Optimizing Cariprazine Production: A Technical Analysis of Novel Synthetic Routes for Commercial Scale
The pharmaceutical industry continuously seeks robust manufacturing pathways for complex antipsychotic agents, and the synthesis of Cariprazine represents a critical area of innovation for generic and branded drug manufacturers alike. Patent CN110317182B discloses a groundbreaking preparation method that addresses long-standing inefficiencies in the production of this dopamine D3/D2 partial agonist. By shifting away from thermally unstable intermediates and genotoxic reagents, this technology offers a viable solution for enhancing supply chain reliability and product purity. The core innovation lies in the utilization of a trans-2-(trans-4-(3,3-dimethylureido)cyclohexyl) derivative reacted under specific acid-binding conditions, followed by a controlled reduction step. This technical insight report analyzes the mechanistic advantages and commercial implications of this patent for R&D directors and procurement strategists seeking a reliable Cariprazine supplier.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
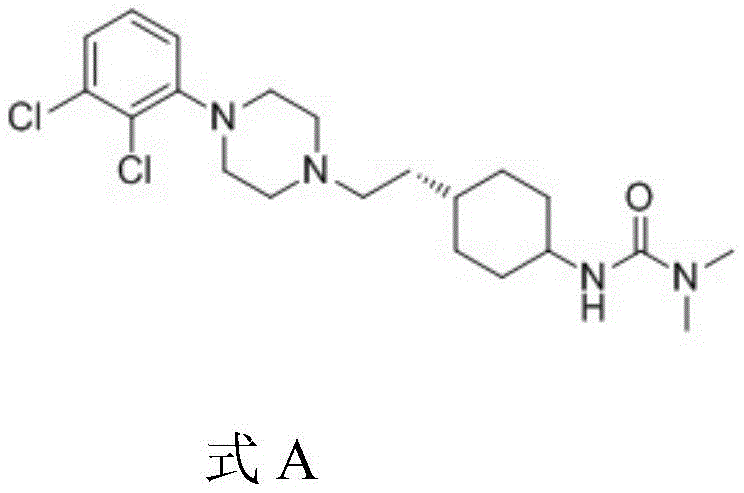
Historically, the industrial synthesis of Cariprazine has relied on routes involving p-toluenesulfonyl chloride to activate intermediate hydroxyl groups, a strategy that introduces significant regulatory and operational risks. The legacy process requires the formation of a tosylate intermediate, such as SM01, which exhibits poor thermal stability and begins to degrade at temperatures as low as 60°C. Consequently, the subsequent nucleophilic substitution with piperazine derivatives must be conducted at elevated temperatures between 80°C and 90°C to drive the reaction to completion, creating a precarious balance between reaction kinetics and intermediate decomposition. This thermal sensitivity results in incomplete conversions and the persistent formation of process-related impurities, capping the coupling yield at a suboptimal 75-80% range. Furthermore, the extended reaction times of 10-12 hours required to achieve even these modest yields impose a heavy burden on reactor occupancy and energy consumption. Most critically from a safety and compliance perspective, the use of tosylating agents generates p-toluenesulfonate esters, which are classified as genotoxic impurities requiring stringent and costly purification protocols to ensure patient safety.
The Novel Approach
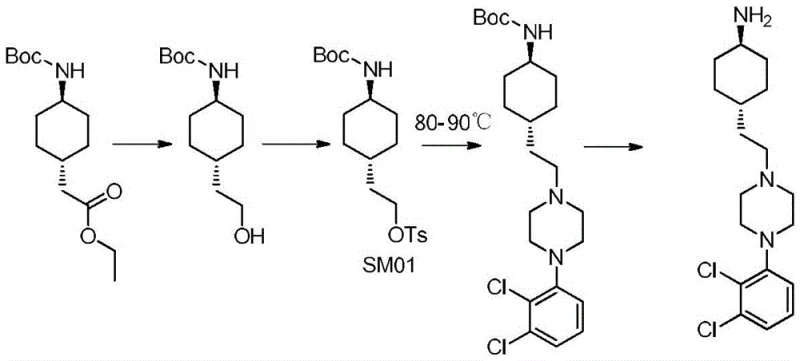
In stark contrast to the thermal constraints of the prior art, the method disclosed in CN110317182B leverages the high reactivity of acyl chloride intermediates to facilitate coupling under mild conditions. By converting the carboxylic acid precursor into an acyl chloride species using reagents like thionyl chloride or oxalyl chloride, the activation energy for the subsequent amide bond formation is significantly lowered. This chemical activation allows the coupling reaction with 1-(2,3-dichlorophenyl)piperazine to proceed efficiently at temperatures between 0°C and 10°C, completely eliminating the risk of thermal degradation associated with the legacy tosylate route. The enhanced reactivity of the acyl chloride group ensures rapid conversion, reducing the reaction time from over 10 hours to merely 0.5-1 hour while simultaneously boosting yields to the 90-95% range. This paradigm shift not only simplifies the process control parameters but also inherently removes the genotoxic tosylate impurity profile, thereby streamlining the downstream purification workflow and reducing the overall environmental footprint of the manufacturing process.
Mechanistic Insights into Acyl Chloride Activation and Amide Reduction
The core of this technological advancement rests on the strategic manipulation of the carbonyl functionality within the cyclohexyl side chain. The process initiates with the hydrolysis of an ethyl ester to form the corresponding carboxylic acid, which is then transformed into a highly electrophilic acyl chloride (Formula III-1). This activation step is crucial because the acyl chloride carbon is significantly more susceptible to nucleophilic attack by the piperazine nitrogen than the corresponding alcohol or tosylate groups used in older methods. The reaction mechanism involves the formation of a tetrahedral intermediate that rapidly collapses to expel the chloride ion, forming the stable amide bond (Formula IV) with high stereochemical fidelity. The use of acid-binding agents such as triethylamine or potassium carbonate in stoichiometric excess ensures the neutralization of the hydrochloric acid byproduct, driving the equilibrium towards product formation and preventing the protonation of the piperazine nucleophile which would otherwise inhibit the reaction. This precise control over the acid-base chemistry is fundamental to achieving the reported high yields and minimizing side reactions.
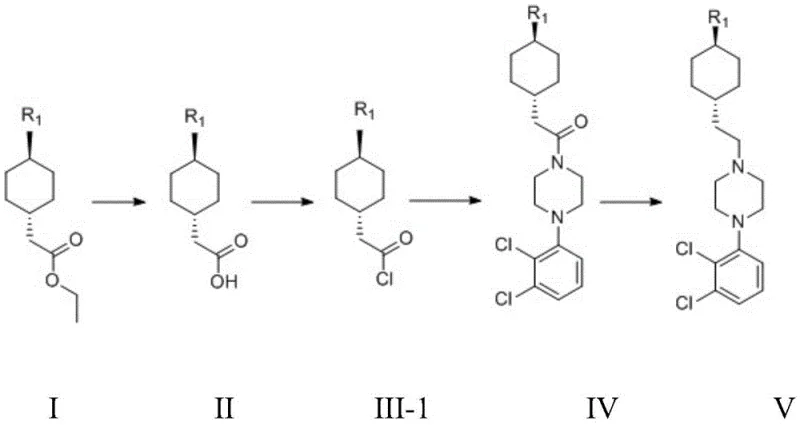
Following the amide coupling, the synthesis proceeds through a critical reduction step to convert the carbonyl group into the required methylene linkage, completing the ethyl bridge between the cyclohexyl and piperazine rings. The patent specifies the use of potent hydride reducing agents such as lithium aluminum hydride (LiAlH4) or DIBAL-H, which are capable of reducing the amide functionality directly to the amine without affecting the sensitive urea moiety or the dichlorophenyl ring. The selection of the reducing agent and solvent system, typically tetrahydrofuran or diethyl ether, is optimized to maintain the integrity of the trans-stereochemistry of the cyclohexyl ring. Impurity control is further enhanced by the absence of sulfonate esters, meaning the primary impurities are likely limited to unreacted starting materials or over-reduction byproducts which are easier to separate via standard crystallization or chromatography. The final salt formation step using hydrochloric acid ensures the production of the stable hydrochloride salt form, which is the marketed pharmaceutical form, with stringent purity specifications suitable for global regulatory submission.
How to Synthesize Cariprazine Efficiently
Implementing this novel synthesis route requires precise adherence to the reaction conditions outlined in the patent to maximize the benefits of the acyl chloride chemistry. The process begins with the preparation of the acyl chloride intermediate under anhydrous conditions to prevent hydrolysis, followed by the immediate addition of the piperazine derivative in the presence of a suitable base. Operators must maintain strict temperature control during the exothermic coupling phase to ensure safety and product quality. The subsequent reduction step demands careful quenching protocols to handle the reactive aluminum species safely. For a detailed breakdown of the specific reagent quantities, solvent volumes, and workup procedures, please refer to the standardized synthesis guide below.
- Hydrolysis of ethyl ester intermediate to form the corresponding carboxylic acid under alkaline conditions.
- Activation of the carboxylic acid using thionyl chloride or oxalyl chloride to generate the reactive acyl chloride species.
- Coupling with 1-(2,3-dichlorophenyl)piperazine followed by reduction of the amide bond to yield the final amine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis pathway translates into tangible operational efficiencies and risk mitigation strategies. The elimination of genotoxic reagents significantly reduces the burden on quality control laboratories, as the testing protocols for p-toluenesulfonate residues are no longer required, thereby accelerating the release of finished goods. The drastic reduction in reaction time from half a day to under an hour per batch increases the throughput capacity of existing manufacturing assets without the need for capital expenditure on new reactors. This intensification of the process allows for more flexible production scheduling and faster response times to market demand fluctuations. Furthermore, the improved yield directly correlates to a reduction in the cost of goods sold, as less raw material is wasted in the form of byproducts or unreacted starting materials. The use of commodity chemicals like thionyl chloride and standard solvents ensures that the supply chain remains resilient against shortages of specialized reagents.
- Cost Reduction in Manufacturing: The transition to the acyl chloride route eliminates the need for expensive purification steps required to remove genotoxic tosylate impurities, leading to substantial cost savings in downstream processing. The higher reaction yield means that less starting material is required to produce the same amount of API, effectively lowering the raw material cost per kilogram. Additionally, the reduced reaction time lowers energy consumption for heating and stirring, contributing to a leaner manufacturing cost structure. These efficiencies compound over large production volumes, offering a competitive pricing advantage in the generic API market.
- Enhanced Supply Chain Reliability: By relying on widely available reagents such as thionyl chloride and potassium carbonate, the manufacturing process is less vulnerable to supply disruptions associated with niche specialty chemicals. The robustness of the reaction conditions, which tolerate mild temperatures and standard solvents, ensures consistent batch-to-batch quality and reduces the risk of production failures due to process deviations. This reliability is critical for maintaining continuous supply to formulation partners and avoiding costly stockouts. The simplified process flow also reduces the lead time for production campaigns, enabling faster replenishment of inventory levels.
- Scalability and Environmental Compliance: The removal of genotoxic impurities simplifies the environmental, health, and safety (EHS) profile of the manufacturing site, reducing the costs associated with hazardous waste disposal and worker safety monitoring. The process is inherently scalable because it avoids the thermal hazards associated with high-temperature tosylation reactions, making it safer to operate at the multi-ton scale. The use of standard organic solvents allows for efficient recovery and recycling systems, further minimizing the environmental footprint. This alignment with green chemistry principles enhances the sustainability credentials of the supply chain, which is increasingly important for corporate social responsibility reporting.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Cariprazine synthesis method. These answers are derived directly from the experimental data and technical disclosures within the patent documentation. They are intended to provide clarity on the feasibility and advantages of this route for potential manufacturing partners and technical evaluators.
Q: How does the novel acyl chloride route improve upon the traditional tosylate method?
A: The novel route eliminates the use of p-toluenesulfonyl chloride, thereby removing the risk of genotoxic p-toluenesulfonate impurities. Additionally, the acyl chloride intermediate is highly reactive, allowing the coupling reaction to proceed at 0-10°C with significantly shorter reaction times compared to the high-temperature requirements of the legacy process.
Q: What are the critical yield improvements observed in the new synthesis pathway?
A: Experimental data from the patent indicates that the coupling step yield improves from approximately 75-80% in the conventional method to 90-95% in the novel acyl chloride method. This substantial increase in efficiency reduces raw material waste and overall production costs.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process utilizes commercially available reagents such as thionyl chloride and standard solvents like dichloromethane and tetrahydrofuran. The reaction conditions are mild (0-10°C for coupling), which enhances safety and controllability during scale-up operations.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cariprazine Supplier
The technical potential of the acyl chloride-mediated synthesis route for Cariprazine represents a significant opportunity for optimizing API production costs and quality. NINGBO INNO PHARMCHEM, as a seasoned CDMO partner, possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this innovative process to fruition. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of managing the specific analytical requirements of complex antipsychotic intermediates. We understand the critical nature of impurity control and yield optimization in the pharmaceutical sector and are prepared to implement this patent-protected methodology with the highest standards of operational excellence.
We invite procurement leaders to engage with our technical procurement team to discuss how this advanced synthesis route can be integrated into your supply chain strategy. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic benefits specific to your volume requirements. We encourage you to contact us to obtain specific COA data and route feasibility assessments that demonstrate our capability to deliver high-purity Cariprazine efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →