Optimizing Ganciclovir Purity: A Strategic Analysis of Advanced Refining Technologies for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust methodologies to enhance the purity profiles of critical antiviral agents, and patent CN102627643A presents a significant advancement in the refining technology for ganciclovir. This specific intellectual property addresses the persistent challenge of controlling guanine impurities, which are structurally similar byproducts that often persist through standard synthesis routes. The disclosed method introduces a strategic combination of lower fatty acid dissolution and inorganic alkali treatment to achieve a guanine impurity content below 0.50 percent without the need for repetitive crystallization operations. For R&D directors and procurement specialists, this represents a pivotal shift from labor-intensive purification protocols to a more streamlined, energy-efficient workflow. By leveraging the differential solubility characteristics of the target compound versus its impurities in acidic media, the process ensures high yield retention while drastically cutting down processing time. This technical breakthrough not only optimizes the chemical quality but also aligns perfectly with the demands for cost reduction in pharmaceutical manufacturing, offering a viable pathway for reliable ganciclovir supplier partnerships aiming for commercial excellence.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the purification of ganciclovir has been plagued by inefficient protocols that rely heavily on repeated recrystallization and activated charcoal adsorption to remove stubborn impurities. Traditional bibliographical methods often necessitate dissolving the crude bulk material in hot water, followed by multiple cycles of charcoal treatment and cooling crystallization to gradually lower impurity levels. In many documented cases, achieving a qualified purity profile requires repeating these thermal and adsorption operations more than eight times, which creates a substantial bottleneck in production throughput. This iterative process not only consumes excessive amounts of thermal energy and solvent but also leads to significant product loss during each filtration and transfer step, ultimately depressing the overall yield. Furthermore, the reliance on charcoal introduces risks of particulate contamination and requires additional validation steps to ensure no adsorbent residues remain in the final active pharmaceutical ingredient. For supply chain heads, these inefficiencies translate into longer lead times for high-purity pharmaceutical intermediates and increased operational expenditures that erode profit margins in a competitive market.
The Novel Approach
In stark contrast to the cumbersome legacy techniques, the novel approach detailed in the patent utilizes a sophisticated acid-base refinement strategy that simplifies the workflow into two primary stages. The process begins by dissolving the crude ganciclovir in a heated lower fatty acid aqueous solution, such as formic acid or acetic acid, which selectively solubilizes the target molecule while leaving certain insolubles behind for immediate removal. Following filtration, the solution undergoes controlled cooling to induce crystallization, after which the solid is treated with a specific concentration of inorganic alkali solution to hydrolyze any residual acylated impurities. This chemical modification step is crucial as it converts trace ganciclovir acylates back into the free base form, ensuring that the final product meets stringent purity specifications without needing further recursive purification. The elimination of repetitive charcoal adsorption and the reduction of crystallization cycles result in a drastically simplified operation that preserves yield and minimizes waste generation. This methodology offers a clear advantage for commercial scale-up of complex pharmaceutical intermediates, providing a robust framework for manufacturers to enhance their production capabilities.
Mechanistic Insights into Acid-Base Refinement and Impurity Control
The core mechanism driving the success of this purification technique lies in the exploitation of solubility differentials between ganciclovir and its primary structural analog, guanine, within an organic acid medium. Guanine exhibits relatively higher solubility in aqueous solutions of organic acids compared to the target nucleoside under specific thermal conditions, allowing for a preliminary separation during the hot filtration stage. 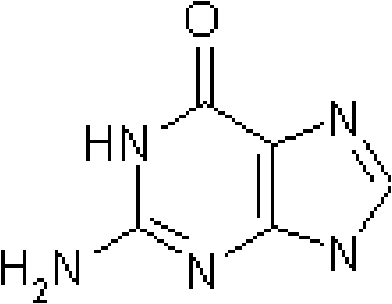 As illustrated by the chemical structure of the guanine impurity, its planar heterocyclic nature interacts differently with the acidic solvent matrix than the sugar-modified ganciclovir molecule. By maintaining the solution at reflux temperatures with formic acid, the process ensures that the maximum amount of ganciclovir remains in the solution phase while insoluble mechanical impurities are filtered out. Upon cooling, the solubility of ganciclovir decreases sharply, prompting it to precipitate out of the solution while a significant portion of the soluble guanine remains in the mother liquor. This physical separation is the first critical barrier against contamination, setting the stage for the subsequent chemical treatment that guarantees the final quality standards required by regulatory bodies.
As illustrated by the chemical structure of the guanine impurity, its planar heterocyclic nature interacts differently with the acidic solvent matrix than the sugar-modified ganciclovir molecule. By maintaining the solution at reflux temperatures with formic acid, the process ensures that the maximum amount of ganciclovir remains in the solution phase while insoluble mechanical impurities are filtered out. Upon cooling, the solubility of ganciclovir decreases sharply, prompting it to precipitate out of the solution while a significant portion of the soluble guanine remains in the mother liquor. This physical separation is the first critical barrier against contamination, setting the stage for the subsequent chemical treatment that guarantees the final quality standards required by regulatory bodies.
Following the initial crystallization, the second mechanistic pillar involves the treatment of the solid cake with an inorganic alkali solution, typically sodium hydroxide or potassium hydroxide. During the synthesis of ganciclovir, side reactions often generate acylated derivatives where the hydroxyl groups are protected or modified; these derivatives can persist as impurities if not properly addressed. The alkaline treatment serves to hydrolyze these ester linkages, converting any ganciclovir acylates back into the desired ganciclovir free base, thereby recovering yield that would otherwise be lost as impurity. After this hydrolysis step, the pH is carefully adjusted to neutrality using hydrochloric acid, which induces a final crystallization event that further purifies the lattice structure of the solid. This dual-action mechanism of physical solubility separation followed by chemical conversion ensures that the guanine foreign matter content is reduced to below 0.50 percent consistently. For technical teams, understanding this interplay between solubility physics and reaction chemistry is essential for troubleshooting and optimizing the process during technology transfer.
How to Synthesize Ganciclovir Efficiently
Implementing this refined synthesis route requires precise control over reaction parameters to maximize the efficiency of impurity removal while maintaining high recovery rates. The process is designed to be operationally simple, utilizing common laboratory and industrial reagents that are readily available in the global chemical supply chain. Operators must carefully monitor the temperature during the dissolution phase to ensure complete solubilization without degrading the thermally sensitive nucleoside structure. The subsequent addition of activated carbon, though used sparingly compared to traditional methods, still plays a role in decolorizing the solution before the critical cooling crystallization step. Detailed standard operating procedures regarding the concentration of the formic acid and the stoichiometry of the neutralization step are vital for reproducibility. The following guide outlines the standardized synthesis steps derived from the patent embodiments to assist process engineers in replicating these results.
- Dissolve crude ganciclovir in a heated lower fatty acid aqueous solution (e.g., formic acid) and filter off insoluble substances.
- Cool the filtrate to induce crystallization, then suction-filter to collect the solid product.
- Treat the solid with an inorganic alkali solution, adjust to neutral pH, and perform a final crystallization to obtain high-purity ganciclovir.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this advanced refining method offers substantial benefits that extend far beyond mere technical compliance, directly impacting the bottom line and supply chain resilience. By eliminating the need for multiple recrystallization cycles and extensive charcoal treatments, manufacturers can significantly reduce the operational time required per batch, thereby increasing overall plant throughput. This efficiency gain translates into tangible cost savings in utility consumption, particularly in steam and cooling water usage, which are major cost drivers in large-scale chemical processing. Furthermore, the improved yield resulting from the recovery of acylated intermediates means that less raw material is required to produce the same amount of finished goods, effectively lowering the cost of goods sold. For procurement managers, this efficiency allows for more competitive pricing strategies without sacrificing margin, making the supply of high-purity ganciclovir more economically sustainable in the long term.
- Cost Reduction in Manufacturing: The streamlined process eliminates expensive and time-consuming repetitive purification steps, leading to significant operational expenditure savings. By reducing the number of heating and cooling cycles, the energy footprint of the manufacturing process is drastically lowered, contributing to both economic and environmental sustainability goals. Additionally, the higher yield achieved through the hydrolysis of acylated byproducts ensures that raw material costs are optimized, providing a direct financial advantage over conventional methods. These cumulative efficiencies allow for a more robust cost structure that can withstand market fluctuations in raw material pricing.
- Enhanced Supply Chain Reliability: The simplicity of the reagents involved, such as formic acid and sodium hydroxide, ensures that the supply chain is not dependent on exotic or hard-to-source catalysts. This availability reduces the risk of production delays caused by material shortages, ensuring a consistent flow of products to downstream customers. The shorter refining cycle also means that inventory turnover is faster, allowing suppliers to respond more agilely to sudden spikes in demand from pharmaceutical clients. Consequently, partners can rely on a more stable and predictable supply of critical antiviral intermediates, mitigating the risks associated with production bottlenecks.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, utilizing unit operations like filtration and crystallization that are easily adapted from pilot scale to multi-ton production. The reduction in solvent usage and waste generation aligns with increasingly strict environmental regulations, minimizing the burden on waste treatment facilities. By avoiding the use of heavy metal catalysts or complex chromatographic resins, the process simplifies the disposal of chemical waste and reduces the environmental impact of the manufacturing site. This compliance readiness makes the technology an attractive option for companies looking to future-proof their production capabilities against tightening global environmental standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this ganciclovir purification technology. These insights are derived directly from the patent specifications and are intended to clarify the operational advantages for potential partners and stakeholders. Understanding these details is crucial for evaluating the feasibility of integrating this method into existing production lines or for assessing the quality credentials of a potential supplier. The answers provided reflect the consensus on how this technology resolves historical pain points in nucleoside manufacturing.
Q: How does this refining method reduce guanine impurity levels?
A: The method utilizes the differential solubility of guanine in lower fatty acid aqueous solutions compared to ganciclovir, followed by selective hydrolysis and recrystallization to achieve impurity levels below 0.50%.
Q: What are the energy consumption advantages over traditional charcoal adsorption?
A: Unlike traditional methods requiring repeated hot water dissolution and charcoal adsorption cycles, this novel approach eliminates multiple recrystallization steps, significantly shortening the refining cycle and reducing thermal energy requirements.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process avoids complex chromatographic separations and uses common reagents like formic acid and sodium hydroxide, making it highly scalable for commercial manufacturing from 100 kgs to 100 MT annual capacity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ganciclovir Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising results seen in patent literature are faithfully reproduced on an industrial scale. We maintain stringent purity specifications across all our batches, supported by rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify impurity profiles. Our commitment to quality assurance means that every kilogram of ganciclovir we supply meets the exacting standards required for pharmaceutical applications, providing peace of mind to R&D directors and quality assurance teams alike.
We invite you to collaborate with us to optimize your supply chain and leverage these advanced purification technologies for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out to request specific COA data and route feasibility assessments to see how our capabilities align with your strategic goals. By partnering with us, you gain access to a reliable source of high-quality intermediates that can accelerate your drug development timelines and enhance your market competitiveness.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →