Revolutionizing Antifungal API Production: A Deep Dive into Novel Azepine Cyclohexapeptide Synthesis
Revolutionizing Antifungal API Production: A Deep Dive into Novel Azepine Cyclohexapeptide Synthesis
The pharmaceutical landscape for antifungal treatments has long been dominated by echinocandin-class compounds, with Caspofungin standing out as a cornerstone therapy due to its unique mechanism of action and broad-spectrum efficacy. However, the manufacturing of this critical Active Pharmaceutical Ingredient (API) has historically been plagued by complex synthetic routes that hinder scalability and cost-efficiency. Patent CN101648994B introduces a transformative approach to this challenge by disclosing a novel class of azepine cyclohexapeptides and a streamlined preparation method that fundamentally alters the production economics of Caspofungin. This technology shifts the paradigm from a cumbersome, low-yield multi-step process to a concise, high-efficiency pathway centered around a stable intermediate known as Formula 4. For R&D directors and supply chain leaders alike, this represents a significant opportunity to optimize the manufacturing of high-purity pharmaceutical intermediates.
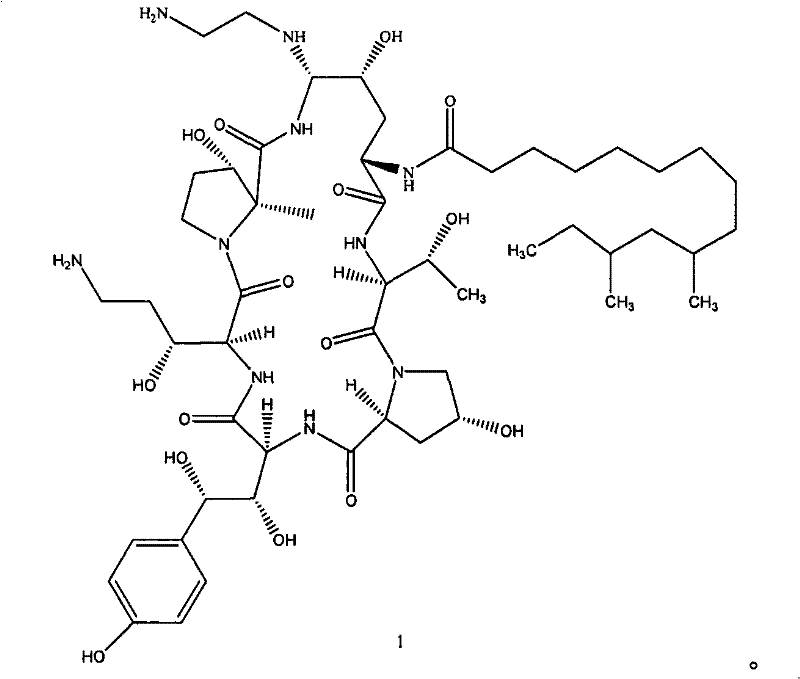
The core innovation lies in the identification and utilization of specific azepine cyclohexapeptide structures that serve as superior precursors to the final drug substance. Unlike previous iterations where intermediates were often unstable oils requiring immediate conversion, the compounds described in this patent, particularly Formula 4, exhibit remarkable stability as solid forms. This characteristic is not merely a chemical curiosity but a pivotal engineering advantage that allows for rigorous quality control, extended storage, and flexible batch scheduling. By decoupling the synthesis into distinct, manageable stages involving thiol substitution and ethylenediamine-mediated cyclization, the process mitigates the risks associated with continuous flow synthesis of unstable species, thereby ensuring a more reliable supply of high-purity API intermediates for global markets.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this technological breakthrough, the industrial synthesis of Caspofungin was largely constrained by the methodologies outlined in patents such as USP 5378804. These conventional routes typically necessitated a minimum of five distinct reaction steps to construct the complex cyclic peptide backbone, a sequence that inherently accumulates impurities and drastically reduces overall throughput. The most glaring deficiency of these legacy processes is the abysmal total reaction yield, which in some documented cases hovers around a mere 6.3%. Such low efficiency translates directly into exorbitant raw material consumption and massive waste generation, creating a bottleneck for cost reduction in pharmaceutical manufacturing. Furthermore, the intermediates generated in these older pathways often lacked significant stereoselectivity, leading to difficult-to-separate diastereomers that compromised the final drug's purity profile and required expensive, resource-intensive purification protocols like preparative HPLC.
The Novel Approach
In stark contrast, the methodology presented in CN101648994B offers a streamlined alternative that collapses the synthetic timeline while simultaneously boosting output. The new approach leverages a fermentation-derived starting material (Formula 2) and subjects it to a highly selective thiol substitution followed by a rapid cyclization with ethylenediamine. This strategy effectively bypasses several tedious protection and deprotection cycles found in traditional syntheses. The result is a robust three-step process that generates stable solid intermediates capable of being purified through simple crystallization or standard column chromatography. This shift from unstable, low-yielding oils to isolable, high-purity solids represents a quantum leap in process chemistry, enabling manufacturers to achieve commercial scale-up of complex pharmaceutical intermediates with greater confidence and reduced operational risk.
Mechanistic Insights into Thiol-Mediated Cyclization and Reduction
The heart of this synthetic innovation is a carefully orchestrated sequence of nucleophilic substitutions and reductive transformations. The process initiates with the reaction of the fermentation product, Formula 2, with a strong leaving group compound, typically a substituted aromatic thiol such as thiophenol. This step is catalyzed by medium-strength acids like trifluoroacetic acid (TFA) or trifluoromethanesulfonic acid, facilitating the displacement of the native leaving group to form the thioether intermediate, Formula 3. Crucially, the patent highlights the optional use of phenylboronic acid during this stage to protect adjacent hydroxyl groups on the tyrosine fragment. This protection strategy is mechanistically vital as it sterically hinders the formation of bis-diphenyl sulfide impurities, a common side reaction that plagues thiol chemistry in peptide systems.
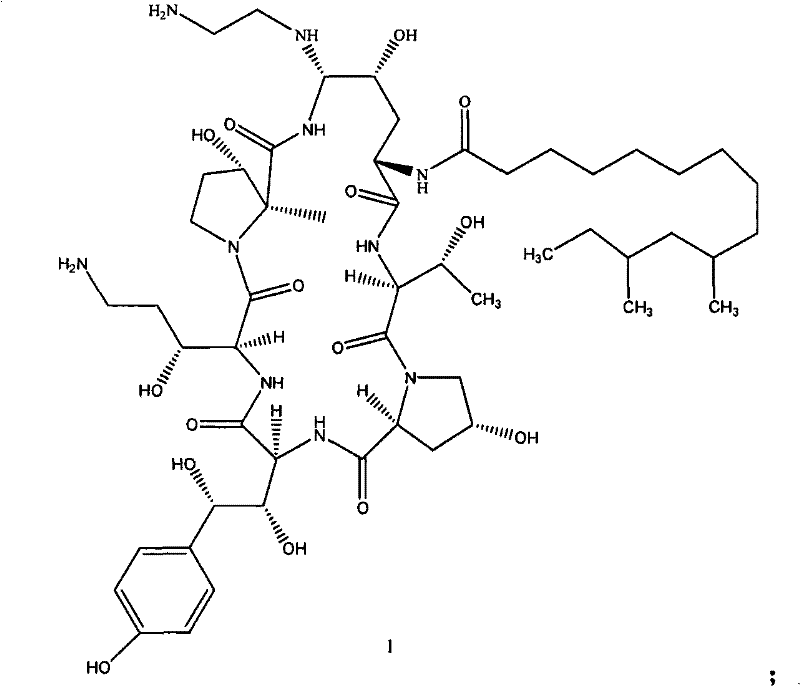
Following the formation of the thioether, the second critical transformation involves the treatment of Formula 3 with ethylenediamine in a polar solvent. This step induces an intramolecular cyclization that constructs the characteristic seven-membered azepine ring, yielding the target intermediate, Formula 4. The mechanism here relies on the nucleophilicity of the diamine to displace the thiol group, closing the macrocycle with high fidelity. Finally, for the production of the active Caspofungin API (Formula 1), the amide functionality within Formula 4 is selectively reduced using a borane complex, such as borane-THF. This reduction is performed under mild conditions (0°C to 10°C) to preserve the stereochemical integrity of the surrounding chiral centers, ensuring that the final product meets the stringent enantiomeric excess requirements demanded by regulatory bodies for antifungal medications.
Impurity control is another pillar of this mechanistic design. The patent explicitly addresses the formation of unwanted diphenyl sulfide byproducts (Formula 5), which can arise if the reaction conditions are not tightly controlled. By optimizing the acid concentration and employing the aforementioned boronic acid protection, the process minimizes these side reactions. Additionally, the use of specific solvents like acetonitrile and precise temperature controls (e.g., maintaining -15°C during the substitution phase) further suppresses degradation pathways. This rigorous attention to mechanistic detail ensures that the crude reaction mixtures are cleaner, reducing the burden on downstream purification units and enhancing the overall mass balance of the manufacturing campaign.
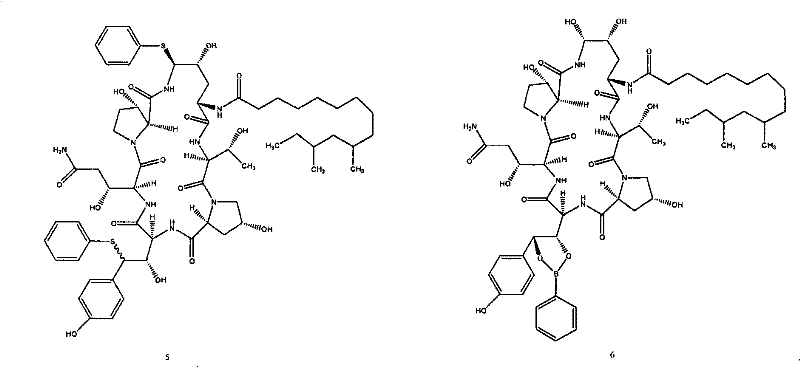
How to Synthesize Azepine Cyclohexapeptides Efficiently
Implementing this synthesis route requires precise adherence to the reaction parameters defined in the patent to maximize yield and purity. The process begins with the dissolution of the fermentation-derived precursor in a suitable solvent like acetonitrile, followed by the controlled addition of thiophenol and an acid catalyst at sub-zero temperatures. Once the thioether intermediate is formed and isolated as a stable solid, it is redissolved and treated with ethylenediamine to effect cyclization. The resulting azepine cyclohexapeptide (Formula 4) can then be isolated via crystallization or chromatography. For those aiming to produce the final Caspofungin API, a subsequent reduction step using borane complexes completes the transformation. The detailed standardized synthesis steps see the guide below.
- React fermentation-derived Compound 2 with a strong leaving group compound (such as thiophenol) in the presence of an acid catalyst to form the thioether intermediate (Compound 3).
- Treat the thioether intermediate with ethylenediamine in a polar solvent to induce cyclization and form the stable azepine cyclohexapeptide (Compound 4).
- Optionally reduce the amide functionality of Compound 4 using a borane complex to obtain the final Caspofungin API (Compound 1).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers compelling strategic advantages that extend beyond simple chemistry. The transition from a five-step, low-yield process to a three-step, high-yield protocol fundamentally reshapes the cost structure of producing echinocandin intermediates. By eliminating two entire reaction steps and their associated workups, the process significantly reduces the consumption of solvents, reagents, and energy. This simplification directly correlates to a substantial reduction in manufacturing costs, allowing for more competitive pricing in the global antifungal market without compromising on quality standards.
- Cost Reduction in Manufacturing: The elimination of complex, low-yielding steps drastically cuts down on raw material waste and processing time. In the legacy process, losing over 90% of material mass across five steps meant that the cost of goods sold was heavily inflated by the sheer volume of starting material required. The new method, with yields reaching up to 90% in individual steps as demonstrated in the patent examples, ensures that a much higher percentage of expensive fermentation broth is converted into saleable product. This efficiency gain removes the need for expensive重金属 removal steps often associated with transition metal catalysts used in other routes, further driving down operational expenditures.
- Enhanced Supply Chain Reliability: One of the most significant logistical benefits is the stability of the intermediates. Traditional peptide synthesis often generates unstable oils that must be processed immediately, creating a rigid, Just-In-Time production schedule that is vulnerable to equipment downtime. In contrast, the ability to isolate Formula 4 as a stable, dry solid allows manufacturers to build inventory buffers. This decoupling of synthesis stages means that if a downstream reduction unit goes offline, the upstream cyclization can continue running, storing the intermediate for later use. This flexibility drastically reduces lead time for high-purity pharmaceutical intermediates and ensures continuous supply even during maintenance windows.
- Scalability and Environmental Compliance: The simplified workflow inherently generates less chemical waste, aligning with modern green chemistry principles and reducing the burden on wastewater treatment facilities. The use of common, recyclable solvents like acetonitrile and methanol, combined with the ability to purify via crystallization rather than solely relying on resource-intensive chromatography, makes this process highly scalable from pilot plant to commercial tonnage. The robust nature of the reaction conditions, which tolerate a broader range of temperatures and concentrations compared to the sensitive legacy routes, ensures that the process can be reliably transferred to large-scale reactors without the fear of runaway reactions or catastrophic yield drops.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These answers are derived directly from the experimental data and claims within CN101648994B, providing a factual basis for evaluating the technology's feasibility for your specific production needs.
Q: How does this new synthesis method improve upon the traditional USP 5378804 process?
A: The traditional method described in USP 5378804 requires five reaction steps with a cumulative yield of only 6.3% and lacks significant stereoselectivity. In contrast, the novel method disclosed in patent CN101648994B reduces the pathway to just three critical steps, utilizes stable solid intermediates that facilitate purification, and achieves significantly higher yields (up to 90% in key steps), making it far more suitable for industrial scale-up.
Q: What role does phenylboronic acid play in the reaction mechanism?
A: Phenylboronic acid acts as a protecting agent for the adjacent hydroxyl groups in the tyrosine fragment during the initial substitution step. This protection is crucial for minimizing the formation of undesirable diphenyl sulfide impurities (Formula 5 compounds), thereby enhancing the overall purity of the intermediate and simplifying downstream processing.
Q: Is the intermediate Compound 4 stable enough for long-term storage and transport?
A: Yes, one of the primary advantages of this invention is the generation of Compound 4 as a stable, dry solid. Unlike unstable oils or gums often encountered in peptide synthesis, this solid intermediate can be easily purified via crystallization or column chromatography, stored, and transported without significant degradation, offering substantial logistical benefits for supply chain management.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Caspofungin Intermediate Supplier
The technological potential of the azepine cyclohexapeptide synthesis route described in CN101648994B is immense, offering a clear path to more affordable and accessible antifungal therapies. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this chemistry to life. Our facility is equipped with state-of-the-art reactors capable of handling the cryogenic conditions (-15°C) and sensitive borane reductions necessary for this process, all while maintaining stringent purity specifications through our rigorous QC labs. We understand that moving from a patent example to a GMP-compliant commercial process requires deep expertise in process safety and impurity profiling, areas where our technical team excels.
We invite you to explore how this optimized synthesis can transform your supply chain economics. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data from our pilot runs and comprehensive route feasibility assessments to demonstrate how we can become your trusted partner in delivering high-quality Caspofungin intermediates to the global market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →