Advanced Palladium-Catalyzed Carbonylation for Scalable N-Acyl Indole Manufacturing
Advanced Palladium-Catalyzed Carbonylation for Scalable N-Acyl Indole Manufacturing
The structural motif of indole serves as a foundational backbone in medicinal chemistry, appearing in a vast array of bioactive molecules ranging from anti-inflammatory agents like Indomethacin to potent anti-tumor compounds. As depicted in the overview of indole-based pharmaceuticals, the demand for efficient synthetic routes to functionalized indoles is critical for modern drug discovery pipelines. 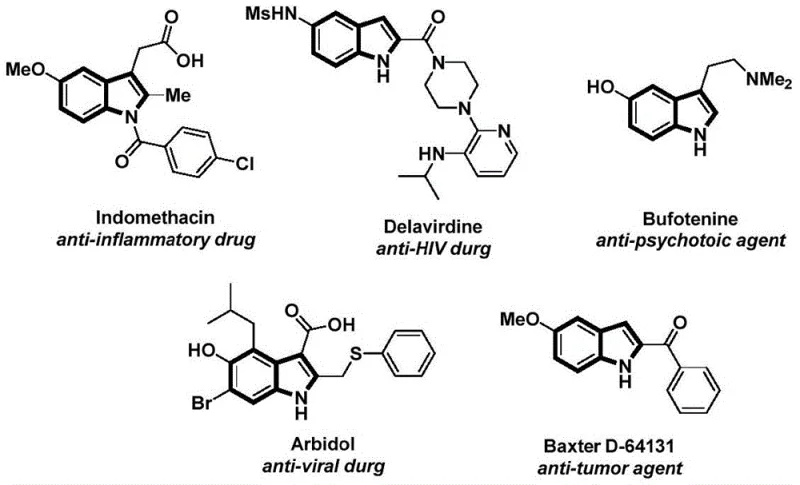 Recent advancements detailed in patent CN112898192B introduce a transformative preparation method for N-acyl indole compounds that addresses long-standing challenges in carbonylation chemistry. This novel approach leverages a palladium-catalyzed cascade reaction utilizing 2-alkynyl aniline and aryl iodides, offering a robust alternative to traditional methods that often suffer from harsh conditions or limited substrate scope. For R&D directors and procurement specialists alike, this technology represents a significant leap forward in accessing high-value pharmaceutical intermediates with improved operational simplicity and cost-efficiency.
Recent advancements detailed in patent CN112898192B introduce a transformative preparation method for N-acyl indole compounds that addresses long-standing challenges in carbonylation chemistry. This novel approach leverages a palladium-catalyzed cascade reaction utilizing 2-alkynyl aniline and aryl iodides, offering a robust alternative to traditional methods that often suffer from harsh conditions or limited substrate scope. For R&D directors and procurement specialists alike, this technology represents a significant leap forward in accessing high-value pharmaceutical intermediates with improved operational simplicity and cost-efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of N-acyl indoles via carbonylation has been fraught with significant technical and safety hurdles that impede large-scale manufacturing. Traditional protocols frequently rely on the direct use of carbon monoxide gas, which poses severe safety risks due to its high toxicity and flammability, necessitating specialized high-pressure equipment and rigorous safety protocols that drive up capital expenditure. Furthermore, conventional palladium-catalyzed carbonylations often require expensive and air-sensitive ligands to maintain catalytic activity, leading to inconsistent yields and difficult purification processes that generate substantial metal waste. The narrow substrate tolerance of older methods frequently results in poor conversion rates when electron-rich or sterically hindered substituents are present, forcing chemists to resort to multi-step synthetic sequences that drastically increase lead times and overall production costs. These inefficiencies create bottlenecks in the supply chain, making it difficult to secure reliable quantities of high-purity intermediates for clinical trials and commercial drug production.
The Novel Approach
The methodology disclosed in the patent overcomes these barriers by employing phenol 1,3,5-tricarboxylate (TFBen) as a safe, solid carbon monoxide surrogate, effectively eliminating the need for handling hazardous CO gas cylinders. This innovative strategy allows the reaction to proceed under mild atmospheric pressure conditions at a moderate temperature of 60°C, significantly reducing energy consumption and equipment requirements. By utilizing tetrakis(triphenylphosphine)palladium as a robust catalyst system alongside potassium carbonate as a base, the process achieves high conversion rates across a broad spectrum of substrates, including those with sensitive functional groups like halogens and alkoxy moieties. The integration of a silver oxide-mediated cyclization step within the same pot streamlines the workflow, transforming what was once a multi-step ordeal into a concise, efficient, and highly scalable operation that is ideally suited for industrial application.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The reaction mechanism proceeds through a sophisticated yet elegant catalytic cycle that ensures high fidelity and yield. Initially, the tetrakis(triphenylphosphine)palladium(0) complex undergoes oxidative addition with the aryl iodide substrate to generate a reactive aryl-palladium intermediate. Subsequently, carbon monoxide, which is thermally released in situ from the decomposition of TFBen, inserts into the palladium-carbon bond to form an acyl-palladium species. This acyl intermediate then reacts with the 2-alkynyl aniline nucleophile, followed by reductive elimination to yield an amide precursor. 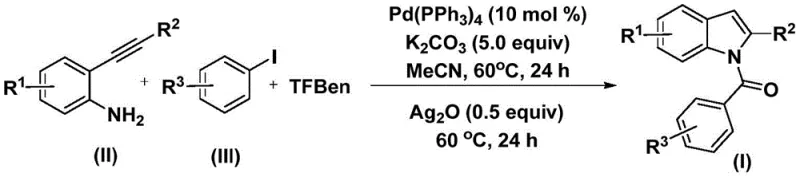 In the second phase of the reaction, the addition of silver oxide acts as a crucial promoter for the intramolecular cyclization of the amide, facilitating the formation of the indole ring system through a dehydrogenative or oxidative pathway that restores aromaticity. This dual-stage mechanism not only maximizes atom economy but also minimizes the formation of side products, ensuring a cleaner reaction profile that simplifies downstream purification.
In the second phase of the reaction, the addition of silver oxide acts as a crucial promoter for the intramolecular cyclization of the amide, facilitating the formation of the indole ring system through a dehydrogenative or oxidative pathway that restores aromaticity. This dual-stage mechanism not only maximizes atom economy but also minimizes the formation of side products, ensuring a cleaner reaction profile that simplifies downstream purification.
From an impurity control perspective, the use of TFBen as a CO source provides a steady, controlled release of carbon monoxide, preventing the rapid accumulation of reactive intermediates that could lead to polymerization or oligomerization byproducts. The choice of acetonitrile as the solvent further enhances selectivity by effectively solubilizing both the organic substrates and the inorganic bases while remaining inert under the reaction conditions. The specific stoichiometry, utilizing 5.0 equivalents of potassium carbonate and 0.5 equivalents of silver oxide, is optimized to drive the equilibrium towards the desired N-acyl indole product while suppressing potential hydrolysis of the acyl group. This precise control over reaction parameters results in a product profile with minimal impurities, reducing the burden on quality control laboratories and ensuring that the final material meets stringent pharmaceutical specifications without extensive recrystallization steps.
How to Synthesize N-Acyl Indole Compounds Efficiently
The operational protocol for this synthesis is designed for maximum reproducibility and ease of execution in both laboratory and pilot plant settings. The process begins by charging a reaction vessel with the palladium catalyst, base, CO source, and substrates in acetonitrile, followed by a controlled heating period to initiate the carbonylation. After the initial transformation is complete, the oxidant is introduced to trigger the cyclization, after which standard workup procedures involving filtration and silica gel chromatography yield the pure product.
- Combine palladium catalyst, potassium carbonate, TFBen (CO source), 2-alkynyl aniline, and aryl iodide in acetonitrile.
- Heat the mixture at 60°C for 24 hours to facilitate the carbonylation and amide formation.
- Add silver oxide to the reaction mixture and continue heating at 60°C for another 24 hours to induce cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers tangible strategic advantages that directly impact the bottom line and operational resilience. By replacing hazardous gaseous reagents with stable solids, the process drastically reduces the regulatory burden and insurance costs associated with handling toxic materials, thereby lowering the total cost of ownership for the manufacturing facility. The reliance on commercially available, off-the-shelf reagents such as aryl iodides and 2-alkynyl anilines ensures a robust and diversified supply base, mitigating the risk of raw material shortages that can plague more exotic synthetic routes. Furthermore, the mild reaction conditions and short overall processing time enhance throughput capacity, allowing manufacturers to respond more agilely to fluctuating market demands without compromising on product quality or delivery schedules.
- Cost Reduction in Manufacturing: The elimination of high-pressure equipment and specialized gas handling infrastructure leads to significant capital expenditure savings, while the use of inexpensive TFBen instead of pressurized CO cylinders reduces variable material costs. The high catalytic efficiency and excellent yields observed across diverse substrates minimize raw material waste, ensuring that every kilogram of input translates effectively into valuable output. Additionally, the simplified one-pot nature of the reaction reduces labor hours and solvent consumption compared to multi-step alternatives, driving down the overall cost per kilogram of the active pharmaceutical ingredient intermediate.
- Enhanced Supply Chain Reliability: Since the key starting materials are commodity chemicals with established global supply chains, the risk of disruption due to single-source dependency is substantially minimized. The robustness of the reaction against variations in substrate electronics means that suppliers can switch between different grades or sources of aryl iodides without needing to re-validate the entire process, providing greater flexibility in sourcing strategies. This stability ensures consistent delivery timelines for downstream customers, fostering stronger long-term partnerships and reducing the need for safety stock inventory.
- Scalability and Environmental Compliance: The absence of toxic gas emissions and the use of recyclable solvents align perfectly with modern green chemistry principles and increasingly strict environmental regulations. The process generates minimal hazardous waste, simplifying disposal protocols and reducing the environmental footprint of the manufacturing site. Its proven scalability from gram to kilogram scales without loss of efficiency makes it an ideal candidate for rapid technology transfer from R&D to commercial production, accelerating time-to-market for new drug candidates.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route, derived directly from the experimental data and scope defined in the patent literature. Understanding these nuances is essential for process chemists aiming to integrate this technology into their existing workflows.
Q: What is the carbon monoxide source in this synthesis?
A: The process utilizes phenol 1,3,5-tricarboxylate (TFBen) as a solid, safe, and efficient carbon monoxide substitute, eliminating the need for hazardous high-pressure CO gas cylinders.
Q: What is the role of Silver Oxide (Ag2O) in the reaction?
A: Silver oxide is added in the second stage of the reaction to promote the intramolecular cyclization of the intermediate amide, converting it into the final N-acyl indole structure.
Q: Does this method tolerate diverse functional groups?
A: Yes, the protocol demonstrates excellent substrate compatibility, successfully accommodating electron-donating groups like methoxy and methyl, as well as electron-withdrawing halogens such as fluorine and chlorine.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Acyl Indole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of your drug development programs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with unwavering consistency. We operate state-of-the-art rigorous QC labs that enforce stringent purity specifications, guaranteeing that every batch of N-acyl indole we deliver is free from detrimental impurities and ready for the next stage of synthesis.
We invite you to collaborate with us to leverage this advanced palladium-catalyzed technology for your specific project needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your target molecule. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your supply chain and accelerate your path to market.