Advanced Mo/Cu Co-Catalyzed Synthesis of 3-Trifluoromethyl-1,2,4-Triazoles for Pharma
Advanced Mo/Cu Co-Catalyzed Synthesis of 3-Trifluoromethyl-1,2,4-Triazoles for Pharma
The landscape of modern pharmaceutical development is increasingly defined by the incorporation of fluorinated heterocycles, which offer superior metabolic stability and bioavailability profiles. A pivotal advancement in this domain is detailed in patent CN113307778A, which discloses a highly efficient preparation method for 3-trifluoromethyl substituted 1,2,4-triazole compounds. These structural motifs are not merely academic curiosities; they are the backbone of critical therapeutics such as Sitagliptin and various anticonvulsant agents, as illustrated in the biological relevance of these scaffolds. For R&D directors and procurement strategists, understanding this novel synthetic route is essential, as it offers a pathway to high-purity intermediates that were previously difficult to access cost-effectively. The technology leverages a sophisticated molybdenum and copper co-catalytic system to achieve what traditional methods could not: a balance of mild conditions, operational simplicity, and broad substrate tolerance.
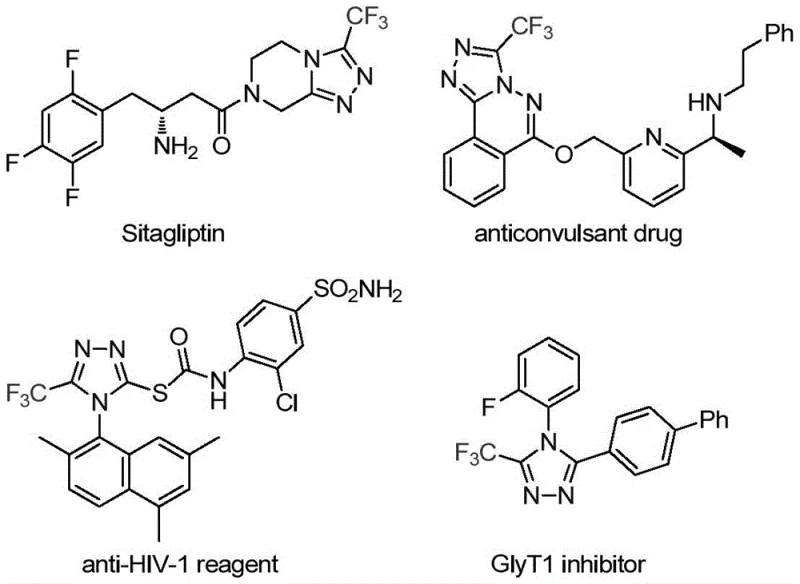
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 1,2,4-triazole ring bearing a trifluoromethyl group has been fraught with synthetic challenges that hinder efficient commercial production. Traditional literature reports predominantly rely on the cyclization of trifluoroacetyl hydrazine with amidine compounds or the hydrazinolysis of trifluoromethyl-substituted 1,2,4-oxazolinones. These legacy pathways often necessitate harsh reaction conditions, involving strong acids or bases that can degrade sensitive functional groups elsewhere in the molecule. Furthermore, alternative multi-component reactions utilizing diazonium salts or trifluorodiazoethane introduce significant safety hazards due to the explosive nature of diazo compounds, creating substantial liability and engineering control costs for manufacturing facilities. The reliance on such dangerous reagents complicates the supply chain, requiring specialized handling protocols and limiting the ability to scale processes safely to multi-ton quantities.
The Novel Approach
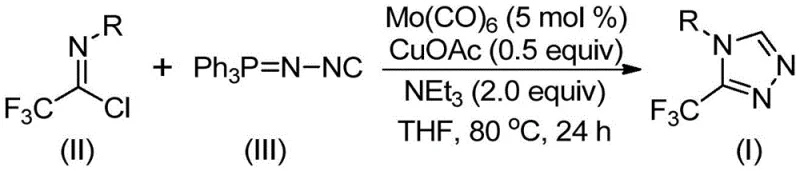
In stark contrast to these hazardous and inefficient legacy routes, the methodology described in CN113307778A introduces a paradigm shift by utilizing trifluoroethylimidoyl chloride and functionalized isonitrile (specifically Ph3P=N-NC) as the primary building blocks. This innovative approach eliminates the need for unstable diazo reagents and harsh hydrazine derivatives, replacing them with stable, commercially available precursors. The reaction proceeds through a concerted cycloaddition mechanism facilitated by the dual metal catalyst system, allowing the transformation to occur at moderate temperatures between 70°C and 90°C. This mildness is a critical advantage for process chemists, as it reduces energy consumption and minimizes the formation of thermal degradation byproducts, thereby simplifying the downstream purification process and enhancing the overall purity profile of the final active pharmaceutical ingredient (API) intermediate.
Mechanistic Insights into Mo/Cu Co-Catalyzed Cycloaddition
The success of this synthetic strategy lies in the intricate interplay between the molybdenum and copper catalytic centers, which orchestrate the bond-forming events with high precision. The mechanism initiates with the activation of the functionalized isonitrile by molybdenum hexacarbonyl, forming a reactive metal-isocyanide complex that primes the carbon-nitrogen triple bond for nucleophilic attack. Simultaneously, the cuprous acetate acts as a Lewis acid promoter, coordinating with the trifluoroethylimidoyl chloride to facilitate a [3+2] cycloaddition event. This cooperative catalysis lowers the activation energy barrier significantly, enabling the formation of the five-membered triazole ring under relatively gentle thermal conditions. The subsequent elimination of triphenylphosphine oxide, driven by the presence of water in the system or during workup, yields the thermodynamically stable 3-trifluoromethyl-1,2,4-triazole core with high regioselectivity.
From an impurity control perspective, this mechanism offers distinct advantages over random thermal cyclizations. The specificity of the metal-ligand interactions ensures that the reaction follows a defined trajectory, minimizing the generation of regioisomers or polymeric side products that often plague uncatalyzed heterocycle synthesis. The use of triethylamine as a base further buffers the reaction environment, scavenging the hydrochloric acid byproduct generated from the imidoyl chloride, which prevents acid-catalyzed decomposition of the sensitive triazole ring. For quality assurance teams, this translates to a cleaner crude reaction profile, reducing the burden on chromatographic purification steps and ensuring that the final material meets the stringent residual solvent and heavy metal specifications required for GMP manufacturing of pharmaceutical intermediates.
How to Synthesize 3-Trifluoromethyl-1,2,4-Triazole Efficiently
The operational protocol for this synthesis is designed for robustness and reproducibility, making it an ideal candidate for technology transfer from laboratory to pilot plant. The process begins by charging a reactor with the precise stoichiometric ratios of molybdenum hexacarbonyl, cuprous acetate, and triethylamine in an anhydrous aprotic solvent such as THF. The substrates, trifluoroethylimidoyl chloride and the functionalized isonitrile, are then introduced, and the mixture is heated to the optimal range of 80°C. Maintaining these conditions for approximately 24 hours ensures complete conversion of the starting materials, as evidenced by the high yields reported across a diverse array of substrates in the patent data. Following the reaction period, the workup involves a straightforward filtration to remove metal salts, followed by adsorption onto silica gel and standard column chromatography to isolate the pure product.
- Combine molybdenum hexacarbonyl (5 mol%), cuprous acetate (0.5 equiv), triethylamine (2.0 equiv), trifluoroethylimidoyl chloride, and functionalized isonitrile in THF solvent.
- Heat the reaction mixture to 70-90°C and maintain stirring for 18 to 30 hours to ensure complete conversion.
- Filter the mixture, adsorb onto silica gel, and purify via column chromatography to isolate the high-purity triazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route represents a strategic opportunity to optimize cost structures and mitigate supply risks associated with complex heterocyclic intermediates. The economic viability of this process is anchored in the use of commodity-grade starting materials; trifluoroethylimidoyl chloride and the requisite amines for isonitrile generation are widely available from global chemical suppliers, eliminating dependence on single-source specialty vendors. This abundance of raw materials fosters a competitive pricing environment, allowing for significant cost reduction in pharmaceutical intermediate manufacturing without compromising on quality. Furthermore, the elimination of hazardous diazo reagents removes the need for expensive blast-proof infrastructure and specialized waste disposal contracts, indirectly lowering the overhead costs associated with production.
- Cost Reduction in Manufacturing: The implementation of this Mo/Cu co-catalyzed system drastically simplifies the production workflow by consolidating multiple synthetic steps into a single pot operation. By avoiding the isolation of unstable intermediates and utilizing cheap catalysts like cuprous acetate, the overall material cost per kilogram of product is substantially decreased. Additionally, the high atom economy of the cycloaddition reaction minimizes waste generation, aligning with green chemistry principles that are increasingly mandated by regulatory bodies and corporate sustainability goals.
- Enhanced Supply Chain Reliability: The robustness of this chemistry against variations in substrate electronics means that a single manufacturing line can produce a wide library of analogues simply by swapping the aromatic amine precursor. This flexibility allows suppliers to respond rapidly to changing R&D demands, reducing lead time for high-purity pharmaceutical intermediates. The stability of the reagents also permits longer storage times and easier logistics, ensuring continuous supply continuity even during global shipping disruptions.
- Scalability and Environmental Compliance: The mild reaction temperatures (70-90°C) and the use of common solvents like THF make this process inherently scalable from gram to multi-ton quantities without requiring exotic high-pressure or cryogenic equipment. The simplified post-treatment procedure, which avoids complex aqueous extractions of hazardous byproducts, streamlines the effluent treatment process. This ease of scale-up ensures that commercial production can meet market demand efficiently while maintaining strict adherence to environmental discharge regulations.
Frequently Asked Questions (FAQ)
To assist technical decision-makers in evaluating this technology for their specific pipelines, we have compiled answers to common inquiries regarding the operational parameters and scope of this synthesis. The following insights are derived directly from the experimental data and claims within the patent documentation, providing a transparent view of the method's capabilities and limitations. Understanding these technical nuances is critical for assessing the feasibility of integrating this route into existing manufacturing portfolios.
Q: What represents the key catalytic advantage of this synthesis method?
A: The method utilizes a dual catalytic system involving molybdenum hexacarbonyl and cuprous acetate, which allows for mild reaction conditions (70-90°C) and high efficiency compared to traditional harsh cyclization methods.
Q: What is the substrate scope for the R group in this triazole synthesis?
A: The process demonstrates excellent tolerance for various substituents, including substituted or unsubstituted aryl groups (with methyl, methoxy, halogen, or nitro groups) and phenethyl groups, allowing for diverse molecular design.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the patent explicitly states that the method can be expanded to gram-level reactions and beyond, utilizing cheap and commercially available starting materials, making it highly viable for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of novel therapeutics depends on the reliability and quality of the underlying chemical supply chain. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 3-trifluoromethyl-1,2,4-triazole intermediate meets the exacting standards required for global regulatory submissions. We are committed to being a reliable pharmaceutical intermediate supplier that prioritizes both technical excellence and commercial partnership.
We invite you to engage with our technical team to explore how this advanced Mo/Cu co-catalyzed technology can be tailored to your specific drug development needs. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits of switching to this greener, more efficient synthetic route. Please contact our technical procurement team today to obtain specific COA data for our catalog compounds and to discuss detailed route feasibility assessments for your proprietary targets.