Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Applications
Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing nitrogen-containing heterocycles, particularly the 1,2,4-triazole scaffold, which serves as a critical pharmacophore in numerous blockbuster drugs. Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in efficiency and environmental compliance. This innovation is particularly relevant for the synthesis of complex molecules such as sitagliptin, maraviroc, and deferasirox, where the introduction of a trifluoromethyl group significantly enhances metabolic stability and lipophilicity. The disclosed technology represents a paradigm shift from traditional transition-metal catalysis to a more sustainable, iodine-promoted organic transformation.
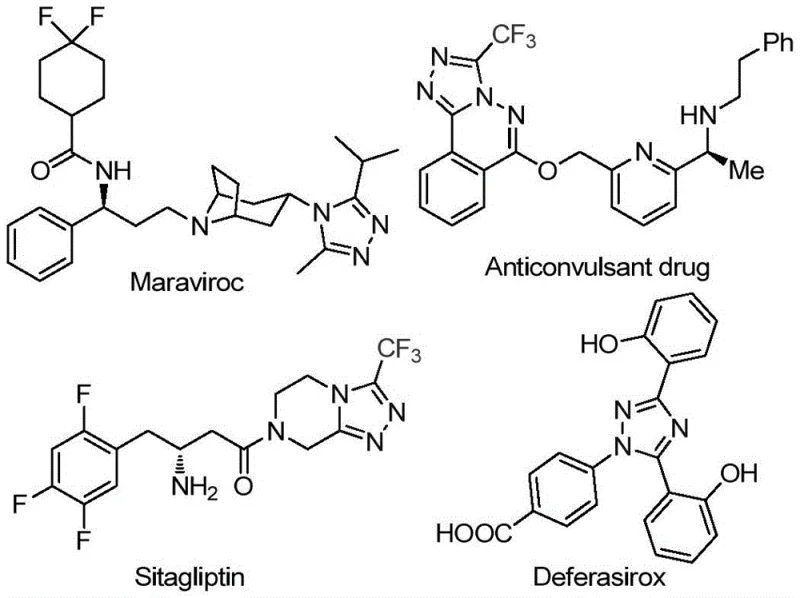
The strategic importance of this patent lies in its ability to construct the triazole core while simultaneously installing a trifluoromethyl group and an acyl functionality in a single operational sequence. For R&D directors overseeing pipeline development, this offers a versatile platform for generating diverse libraries of bioactive candidates. The method circumvents the need for pre-functionalized building blocks that are often prohibitively expensive or unstable. By leveraging a tandem reaction sequence involving iodination, oxidation, and cyclization, the process achieves high atom economy and operational simplicity. This technical advancement positions the technology as a cornerstone for the next generation of high-purity pharmaceutical intermediates, enabling faster lead optimization cycles and more efficient process chemistry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles has relied heavily on transition metal catalysts, such as copper or palladium complexes, to facilitate C-N bond formation and ring closure. These conventional approaches often suffer from significant drawbacks, including the requirement for stringent anhydrous and oxygen-free conditions, which drastically increase operational complexity and infrastructure costs. Furthermore, the use of heavy metal catalysts introduces severe challenges in downstream processing, necessitating rigorous purification steps to meet strict residual metal limits imposed by regulatory bodies like the FDA and EMA. The removal of trace metals often requires specialized scavengers or repeated chromatography, leading to substantial yield losses and increased waste generation. Additionally, many traditional routes utilize expensive or toxic reagents that limit the scalability of the process for commercial API manufacturing.
The Novel Approach
In stark contrast, the methodology described in CN113105402B employs a metal-free strategy driven by elemental iodine and dimethyl sulfoxide (DMSO). This novel approach utilizes a cascade reaction mechanism where aryl ethanones undergo in situ iodination and Kornblum oxidation to generate reactive alpha-dicarbonyl intermediates. These intermediates then condense with trifluoroacetimidoyl hydrazides to form the triazole ring. The elimination of transition metals not only reduces raw material costs but also simplifies the workup procedure to basic filtration and standard chromatography. The reaction proceeds efficiently under ambient atmospheric conditions, removing the need for expensive inert gas manifolds or glovebox techniques. This robustness makes the process highly attractive for commercial scale-up of complex pharmaceutical intermediates, offering a streamlined path from benchtop discovery to pilot plant production.
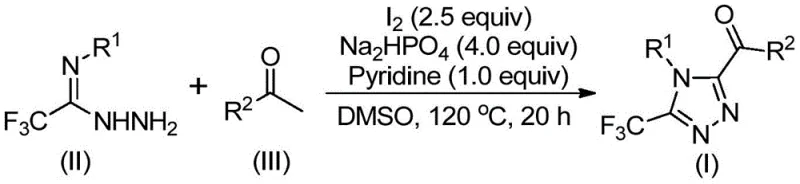
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of oxidative functionalization and cyclocondensation. Initially, the aryl ethanone substrate reacts with elemental iodine in DMSO at elevated temperatures (90-110°C). This step likely involves alpha-iodination followed by nucleophilic substitution by DMSO, leading to an alpha-keto sulfonium species which hydrolyzes or rearranges to an alpha-dicarbonyl compound (aryl glyoxal equivalent). This oxidative step is critical as it activates the methyl group of the ketone for subsequent nucleophilic attack. Upon the addition of the trifluoroacetimidoyl hydrazide, base (Na2HPO4), and pyridine, a condensation reaction occurs to form a hydrazone intermediate. The presence of the electron-withdrawing trifluoromethyl group on the hydrazide enhances the electrophilicity of the imine carbon, facilitating the initial nucleophilic attack by the hydrazine nitrogen.
Following the formation of the hydrazone, the system undergoes an intramolecular cyclization promoted by the continued presence of iodine and base at higher temperatures (110-130°C). This final ring-closing step constructs the 1,2,4-triazole core with high regioselectivity. The tolerance of this mechanism towards various functional groups is exceptional, as evidenced by the successful synthesis of derivatives bearing electron-donating groups (methoxy, methyl) and electron-withdrawing groups (chloro, fluoro, trifluoromethyl) on both the N-aryl and C-acyl moieties. The ability to accommodate heteroaromatic rings, such as furan, further underscores the versatility of this chemistry. For process chemists, understanding this mechanism is vital for troubleshooting and optimizing reaction parameters to minimize impurities such as unreacted starting materials or over-oxidized byproducts.
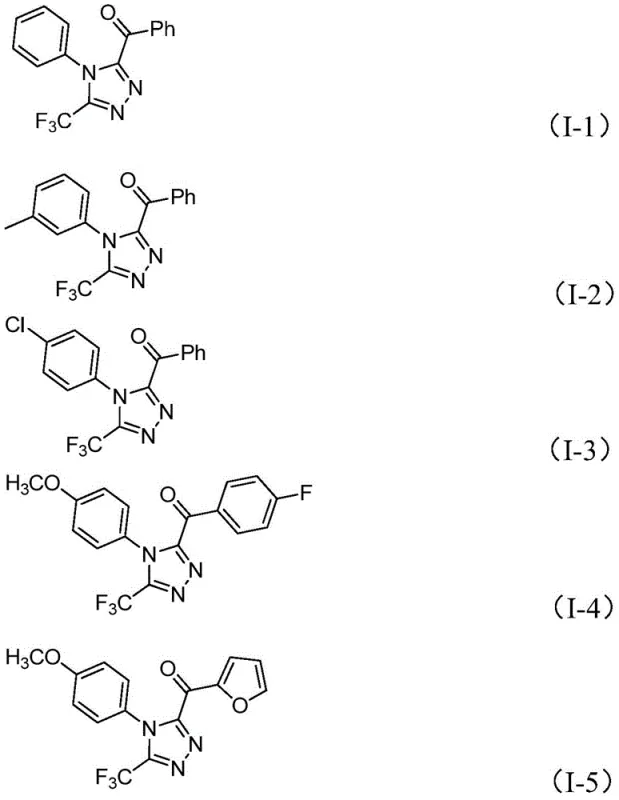
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
Executing this synthesis requires careful attention to the two-stage thermal profile and reagent stoichiometry to ensure maximum conversion and purity. The process begins with the dissolution of the aryl ethanone and a portion of elemental iodine in DMSO, followed by heating to initiate the oxidation phase. After the initial period, the remaining reagents including the hydrazide, base, and pyridine are introduced to drive the cyclization. The detailed standardized synthesis steps, including precise molar ratios and workup procedures, are outlined below to ensure reproducibility and safety in your laboratory operations.
- Stage 1 Oxidation: Dissolve aryl ethanone and elemental iodine in DMSO, heating the mixture to 90-110°C for 4-6 hours to facilitate initial iodination and oxidation.
- Stage 2 Cyclization: Introduce additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroacetimidoyl hydrazide to the reaction vessel.
- Final Conversion: Raise the temperature to 110-130°C and maintain for 12-20 hours to complete the cyclization, followed by filtration and chromatographic purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers transformative advantages that directly impact the bottom line and operational resilience. The shift away from precious metal catalysts to commodity chemicals like iodine and DMSO results in a drastic reduction in raw material expenditure. Moreover, the simplified purification protocol reduces the consumption of silica gel and solvents during the workup phase, contributing to lower overall manufacturing costs. The use of widely available starting materials, such as substituted acetophenones and commercially sourced hydrazides, ensures a stable and diversified supply base, mitigating the risk of shortages that often plague specialized catalytic reagents.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts such as palladium or copper removes a significant cost driver from the bill of materials. Furthermore, avoiding the need for specialized metal scavengers and the associated extensive purification steps leads to substantial savings in processing time and consumables. The operational simplicity allows for the use of standard glass-lined reactors without the need for exotic metallurgy required for corrosive metal salts, thereby reducing capital expenditure and maintenance costs associated with equipment degradation.
- Enhanced Supply Chain Reliability: The reliance on bulk commodity chemicals like aryl ketones and elemental iodine ensures that the supply chain is robust and less susceptible to geopolitical disruptions or vendor bottlenecks. Unlike specialized ligands or organometallic complexes that may have long lead times and limited suppliers, the precursors for this reaction are produced globally at massive scales. This availability facilitates reducing lead time for high-purity pharmaceutical intermediates, allowing for more agile response to market demands and faster time-to-market for new drug formulations.
- Scalability and Environmental Compliance: The process operates under ambient atmospheric conditions, eliminating the energy-intensive requirements for maintaining strict inert atmospheres (nitrogen or argon) throughout the reaction. This feature significantly lowers energy consumption and simplifies the engineering controls needed for scale-up. Additionally, the absence of heavy metals simplifies waste stream management, reducing the environmental burden and costs associated with hazardous waste disposal. The method's proven scalability to gram levels in the patent suggests a straightforward path to kilogram and ton-scale production, ensuring supply continuity for commercial API manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the practical application of this method in an industrial setting.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the patented process (CN113105402B) utilizes elemental iodine as a non-metallic promoter, completely eliminating the need for costly palladium or copper catalysts and simplifying downstream purification.
Q: What are the critical reaction conditions for optimal yield?
A: The process requires a two-step thermal profile in DMSO solvent: an initial heating at 90-110°C for oxidation, followed by a higher temperature phase at 110-130°C for 12-20 hours to drive the cyclization to completion.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the method is designed for scalability as it operates under ambient atmospheric conditions without stringent anhydrous or oxygen-free requirements, using cheap and readily available commodity starting materials.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic methodologies play in the success of modern drug development programs. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and compliant. We are committed to delivering stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of 3,4,5-trisubstituted 1,2,4-triazole intermediate meets the highest global standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this advanced iodine-promoted technology for your specific project needs. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how this metal-free route can optimize your budget. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, and let us partner with you to accelerate your pipeline with reliable, high-quality chemical solutions.