Scalable Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Advanced Drug Discovery
Scalable Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Advanced Drug Discovery
The landscape of heterocyclic chemistry is constantly evolving, driven by the relentless demand for novel scaffolds that can serve as the backbone for next-generation therapeutics. A significant breakthrough in this domain is documented in patent CN116253692A, which details a robust and efficient preparation method for trifluoromethyl-substituted 1,2,4-triazine compounds. These heterocycles are not merely academic curiosities; they are pivotal structures in medicinal chemistry, known for their diverse biological activities ranging from anticancer and antifungal properties to antimalarial and antihypertensive effects. The strategic incorporation of the trifluoromethyl group further amplifies their pharmacological potential by modulating electronegativity and metabolic stability. For R&D directors and procurement specialists alike, understanding the nuances of this synthesis is crucial for securing a reliable supply of high-quality intermediates.
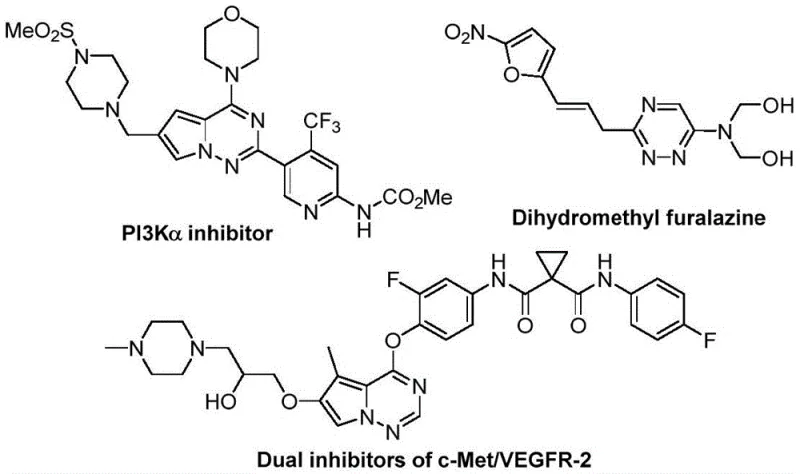
The significance of this technology extends beyond the laboratory bench. As the pharmaceutical industry seeks to optimize its supply chains, the ability to produce complex heterocycles like 1,2,4-triazines through streamlined, cost-effective routes becomes a competitive advantage. The method described in the patent leverages a unique [3+3] cycloaddition strategy that bypasses many of the logistical and financial hurdles associated with traditional synthesis. By utilizing inexpensive inorganic salts and avoiding toxic heavy metals, this process aligns perfectly with the modern principles of green chemistry while delivering the high-purity standards required for active pharmaceutical ingredient (API) manufacturing. This report delves deep into the technical merits and commercial implications of this innovative pathway.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 1,2,4-triazine ring has been a challenging endeavor, often plagued by inefficient protocols that hinder large-scale production. Traditional synthetic routes typically rely on the condensation of amidrazones with 1,2-diketones or alkynes, or multicomponent reactions involving hydrazides and dicarbonyl compounds. While these methods are chemically valid, they frequently suffer from significant drawbacks that impact both cost and throughput. For instance, many conventional processes require the pre-synthesis of complex substrates, adding extra steps and reducing overall atom economy. Furthermore, these reactions often demand harsh conditions, such as elevated temperatures or the use of strong acids and bases, which can lead to the degradation of sensitive functional groups and the formation of difficult-to-remove impurities.
Another critical bottleneck in legacy methods is the reliance on transition metal catalysts or inert atmospheres. The necessity for nitrogen or argon protection increases operational complexity and capital expenditure, as it requires specialized equipment and rigorous safety protocols. Moreover, the presence of heavy metal residues in the final product is a major concern for regulatory compliance, necessitating expensive purification steps to meet strict ppm limits. These factors collectively contribute to longer lead times and higher manufacturing costs, creating friction in the supply chain for key pharmaceutical intermediates. The structural diversity achievable through these older methods is also often limited, restricting the chemical space available for drug discovery teams.
The Novel Approach
In stark contrast to these cumbersome legacy techniques, the methodology outlined in patent CN116253692A introduces a paradigm shift towards simplicity and efficiency. This novel approach utilizes a direct cyclization between chlorohydrazones and trifluoroacetyl sulfur ylides, facilitated by potassium carbonate in a common organic solvent like tetrahydrofuran (THF). The elegance of this reaction lies in its mildness; it proceeds smoothly at room temperature (20-40°C) and, remarkably, under an ambient air atmosphere. This elimination of inert gas requirements drastically simplifies the reactor setup and reduces energy consumption, making it an attractive option for process chemists aiming for scalability.
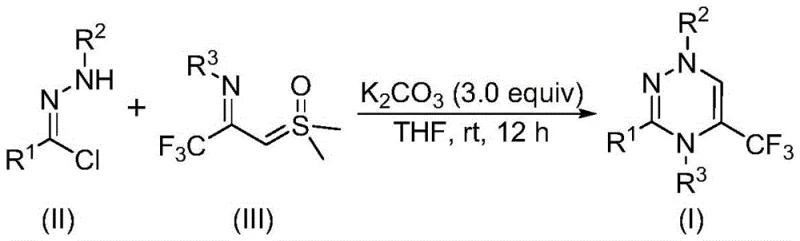
Furthermore, the use of potassium carbonate as a promoter offers distinct advantages over traditional bases. It is inexpensive, odorless, non-toxic, and easy to handle, which enhances workplace safety and reduces waste disposal costs. The reaction demonstrates excellent functional group tolerance, allowing for the introduction of various substituents on the phenyl rings (R1, R2, R3), thereby enabling the rapid generation of diverse libraries of trifluoromethyl-substituted triazines. This flexibility is invaluable for medicinal chemists exploring structure-activity relationships (SAR). By avoiding heavy metal catalysts entirely, the process inherently produces cleaner crude products, minimizing the need for complex downstream purification and ensuring a higher quality output suitable for sensitive biological applications.
Mechanistic Insights into Metal-Free [3+3] Cycloaddition
To fully appreciate the robustness of this synthesis, one must examine the underlying mechanistic pathway. The reaction initiates with the activation of the chlorohydrazone precursor. Under the basic conditions provided by potassium carbonate, the chlorohydrazone undergoes dehydrohalogenation, eliminating a molecule of hydrogen chloride to generate a highly reactive nitrile imine intermediate in situ. This transient species is the key driver of the subsequent cyclization. Simultaneously, the trifluoroacetyl sulfur ylide acts as a dipolarophile, poised for interaction with the nitrile imine. The synergy between these two components facilitates a concerted [3+3] cycloaddition reaction, which constructs the six-membered 1,2,4-triazine core in a single operational step.
Following the cycloaddition, the intermediate undergoes an elimination reaction where a molecule of dimethyl sulfoxide (DMSO) is expelled, aromatizing the ring system to yield the final trifluoromethyl-substituted 1,2,4-triazine product. This mechanism is particularly advantageous because it avoids the formation of stable metal-ligand complexes that often complicate workups in transition-metal catalyzed reactions. From an impurity control perspective, the absence of metal catalysts means there is no risk of metal leaching or the formation of metal-associated byproducts. The primary impurities are likely to be unreacted starting materials or hydrolysis products, which are generally easier to separate via standard chromatographic techniques. This clean reaction profile ensures that the resulting high-purity trifluoromethyl substituted 1,2,4-triazine compounds meet the stringent specifications required for downstream pharmaceutical processing.
How to Synthesize Trifluoromethyl Substituted 1,2,4-Triazine Efficiently
Implementing this synthesis in a practical setting requires adherence to specific operational parameters to maximize yield and reproducibility. The patent data indicates that the stoichiometry of reagents plays a crucial role, with a molar ratio of chlorohydrazone to trifluoroacetyl sulfur ylide to potassium carbonate of approximately 1:2:3 proving optimal. The choice of solvent is also critical; while several aprotic solvents were tested, tetrahydrofuran (THF) demonstrated superior performance in terms of conversion rates and solubility of the reactants. The reaction time typically spans 10 to 14 hours at room temperature, providing a balance between complete conversion and operational efficiency. For detailed procedural specifics regarding reagent addition and workup protocols, please refer to the standardized guide below.
- Combine potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide in an organic solvent such as tetrahydrofuran (THF).
- Stir the reaction mixture at room temperature (20-40°C) under an air atmosphere for 10 to 14 hours to allow the [3+3] cycloaddition to proceed.
- Filter the reaction mixture, mix the filtrate with silica gel, and purify via column chromatography to isolate the high-purity trifluoromethyl substituted 1,2,4-triazine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthesis method represents a strategic opportunity to optimize costs and enhance supply security. The economic benefits are derived not just from the price of raw materials, but from the holistic efficiency of the process. By eliminating the need for expensive transition metal catalysts and specialized inert atmosphere equipment, the overall cost of goods sold (COGS) can be significantly reduced. Additionally, the use of commodity chemicals like potassium carbonate and THF ensures that the supply chain remains resilient against fluctuations in the availability of exotic reagents. This stability is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The most immediate financial impact comes from the removal of heavy metal catalysts. Transition metals such as palladium or rhodium are not only costly to purchase but also require expensive recovery or disposal processes to meet environmental regulations. By replacing these with inexpensive potassium carbonate, the process achieves substantial cost savings in both raw material acquisition and waste management. Furthermore, the ability to run the reaction at room temperature eliminates the energy costs associated with heating or cooling reactors, contributing to a leaner manufacturing budget. The simplified post-treatment, which involves basic filtration and chromatography, reduces labor hours and solvent consumption compared to complex extraction protocols needed for metal removal.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by the reliance on single-source suppliers for specialized reagents. This method mitigates that risk by utilizing starting materials that are commercially available from multiple vendors globally. Chlorohydrazones and sulfur ylides can be synthesized from common precursors like acyl chlorides and hydrazines, which are staple chemicals in the fine chemical industry. This broad availability ensures that production is not held hostage by supply shortages of niche catalysts. Moreover, the robustness of the reaction under air atmosphere means that manufacturing can proceed without the logistical burden of managing bulk nitrogen or argon supplies, further streamlining operations and reducing the risk of downtime due to utility failures.
- Scalability and Environmental Compliance: Scaling chemical processes from the gram level to metric tons often reveals hidden bottlenecks, but this methodology is inherently designed for expansion. The exothermic nature of the reaction is manageable at room temperature, reducing the risk of thermal runaway in large reactors. From an environmental standpoint, the process aligns with green chemistry principles by avoiding toxic heavy metals and generating benign byproducts like DMSO and salts. This reduces the regulatory burden associated with hazardous waste disposal and facilitates easier permitting for new manufacturing facilities. The ability to produce commercial scale-up of complex heterocyclic intermediates with a minimal environmental footprint is a significant competitive differentiator in today's eco-conscious market.
Frequently Asked Questions (FAQ)
Understanding the technical details of a new synthesis route is essential for making informed sourcing decisions. The following questions address common inquiries regarding the feasibility, quality, and application of this trifluoromethyl triazine technology. These answers are derived directly from the experimental data and technical disclosures found in the patent literature, providing a transparent view of the process capabilities.
Q: What are the primary advantages of this new synthesis method over conventional triazine production?
A: Unlike traditional methods that often require harsh conditions, expensive transition metal catalysts, or inert atmospheres, this novel approach utilizes cheap, non-toxic potassium carbonate as a promoter and proceeds efficiently at room temperature in air, significantly simplifying the operational workflow and reducing environmental impact.
Q: How does the incorporation of the trifluoromethyl group benefit the final pharmaceutical application?
A: The introduction of a trifluoromethyl group into the 1,2,4-triazine scaffold markedly improves the molecule's physicochemical properties, including enhanced metabolic stability, increased lipophilicity, and better bioavailability, which are critical parameters for developing potent anticancer and antifungal agents.
Q: Is this synthesis method suitable for large-scale commercial manufacturing?
A: Yes, the process is highly scalable due to its use of readily available starting materials, mild reaction conditions that do not require specialized pressure vessels or cryogenic cooling, and a straightforward post-treatment involving simple filtration and chromatography, making it ideal for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Triazine Supplier
The development of efficient synthetic routes like the one described in CN116253692A underscores the dynamic nature of modern chemical manufacturing. However, translating a patent into a reliable commercial supply requires more than just a recipe; it demands expertise in process optimization, quality control, and scale-up engineering. NINGBO INNO PHARMCHEM stands at the forefront of this industry, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our commitment to excellence ensures that every batch of trifluoromethyl substituted 1,2,4-triazine compounds meets stringent purity specifications, verified through our rigorous QC labs equipped with state-of-the-art analytical instrumentation.
We understand that every project has unique requirements, whether it is a custom derivative for SAR studies or a bulk supply for clinical trials. Our team is ready to collaborate with you to tailor this synthesis to your specific needs, ensuring reducing lead time for high-purity pharmaceutical intermediates while maintaining cost efficiency. We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis. Let us provide you with specific COA data and route feasibility assessments to demonstrate how we can support your drug development pipeline with reliable, high-quality chemical solutions.