Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
The landscape of heterocyclic chemistry is constantly evolving, driven by the demand for more efficient and cost-effective pathways to bioactive scaffolds. Quinazolinone derivatives represent a cornerstone class of nitrogen-containing fused ring systems, ubiquitous in medicinal chemistry due to their profound biological activities ranging from anticancer and anticonvulsant to anti-inflammatory and antifungal properties. As illustrated in the structural diversity of known bioactive molecules such as Afloqualone, Luotonin F, and the TLN inhibitor, the quinazolinone core is a privileged structure in drug discovery. 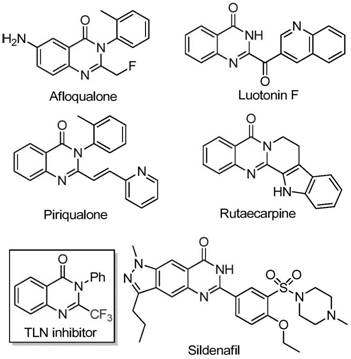 . A recent breakthrough detailed in patent CN111675662B introduces a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. This innovation addresses critical bottlenecks in traditional synthesis by leveraging a cheap iron catalyst system, offering a compelling value proposition for reliable pharmaceutical intermediate suppliers seeking to optimize their manufacturing portfolios.
. A recent breakthrough detailed in patent CN111675662B introduces a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. This innovation addresses critical bottlenecks in traditional synthesis by leveraging a cheap iron catalyst system, offering a compelling value proposition for reliable pharmaceutical intermediate suppliers seeking to optimize their manufacturing portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the introduction of a trifluoromethyl group into the quinazolinone scaffold has been a challenging endeavor, often plagued by significant economic and operational inefficiencies. Conventional literature reports predominantly rely on the cyclization of synthons bearing trifluoromethyl groups, such as trifluoroacetic anhydride or ethyl trifluoroacetate, with substrates like anthranilamide or isatoic anhydride. While chemically feasible, these traditional routes are severely limited by harsh reaction conditions that necessitate rigorous safety protocols and specialized equipment. Furthermore, the starting materials involved, particularly the fluorinated synthons, are frequently expensive and subject to supply chain volatility, which directly impacts the cost of goods sold for the final active pharmaceutical ingredient. The narrow substrate scope of these older methods also restricts the ability of R&D teams to rapidly iterate on molecular designs, slowing down the drug discovery pipeline and increasing the overall time-to-market for new therapeutic candidates.
The Novel Approach
In stark contrast to these legacy methods, the novel approach disclosed in the patent utilizes readily available trifluoroethylimidoyl chloride and isatin derivatives as the foundational building blocks. This strategic shift in synthon selection allows for a series of cyclization reactions catalyzed by inexpensive ferric chloride, a base metal catalyst that drastically reduces raw material costs compared to precious metal alternatives. The process operates under relatively mild conditions initially at 40°C before heating to 120°C, demonstrating excellent functional group tolerance that accommodates a wide variety of substituents including halogens, alkyls, and nitro groups. 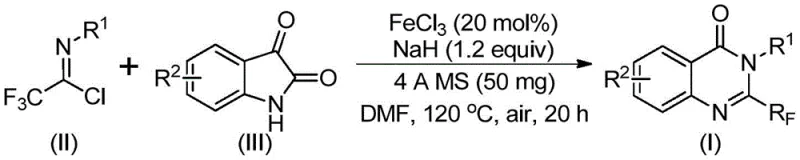 . This robustness not only simplifies the purification process but also ensures high conversion rates, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates where consistency and yield are paramount.
. This robustness not only simplifies the purification process but also ensures high conversion rates, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates where consistency and yield are paramount.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of base-promoted coupling and transition metal catalysis. Initially, the reaction involves the formation of carbon-nitrogen bonds between the trifluoroethylimidoyl chloride and the isatin substrate, facilitated by sodium hydride acting as a strong base promoter. This step generates a trifluoroacetamidine intermediate, which serves as the precursor for the subsequent ring closure. The presence of ferric chloride is critical here, as it catalyzes a decarbonylation and cyclization sequence that isomerizes the intermediate into the final stable quinazolinone architecture. This iron-catalyzed step is particularly noteworthy because it avoids the use of toxic or expensive reagents, aligning with green chemistry principles that are increasingly demanded by global regulatory bodies and corporate sustainability goals.
From an impurity control perspective, the use of 4A molecular sieves in the reaction mixture plays a pivotal role in maintaining high product purity. By sequestering moisture and other small polar byproducts, the molecular sieves prevent hydrolysis of the sensitive imidoyl chloride starting material and suppress side reactions that could lead to difficult-to-remove impurities. The reaction conditions, specifically the two-stage temperature profile (40°C followed by 120°C), are optimized to ensure complete conversion while minimizing thermal degradation. This precise control over the reaction environment results in a clean crude product profile, which significantly reduces the burden on downstream purification processes like column chromatography, thereby enhancing the overall process mass intensity and reducing solvent waste generation.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for reproducing these high-value intermediates with consistent quality. The procedure begins with the careful addition of ferric chloride and sodium hydride to a reaction vessel containing the organic solvent, typically DMF, along with the molecular sieves. The specific molar ratios are critical, with a preferred ratio of trifluoroethylimidoyl chloride to isatin to ferric chloride being approximately 1.2:1:0.2, ensuring that the limiting reagent is fully consumed. Detailed standardized synthetic steps see the guide below for exact parameters regarding stirring times and temperature ramps which are essential for maximizing yield and reproducibility in a GMP environment.
- Mix ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in DMF solvent.
- Stir the reaction mixture at 40°C for approximately 10 hours to initiate the coupling, then heat to 120°C.
- Maintain the reaction at 120°C for 20 hours under air atmosphere, then filter and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method translates into tangible strategic advantages beyond mere chemical curiosity. The shift from expensive fluorinated anhydrides to commodity chemicals like isatin and aromatic amines fundamentally alters the cost structure of the supply chain. By utilizing ferric chloride, a ubiquitous and low-cost industrial chemical, manufacturers can eliminate the dependency on volatile markets for specialized fluorinating agents, leading to substantial cost savings in pharmaceutical intermediates manufacturing. This stability in raw material sourcing ensures that production schedules remain uninterrupted, even during periods of global supply chain disruption, providing a reliable buffer against market fluctuations.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts and expensive trifluoroacetylating agents results in a drastic simplification of the bill of materials. Since ferric chloride is significantly cheaper than palladium or rhodium alternatives often used in C-H activation or cross-coupling, the direct material cost per kilogram of product is markedly lower. Furthermore, the high atom economy of the cyclization reaction minimizes waste disposal costs, contributing to a leaner and more profitable production model without compromising on the quality or purity of the final API intermediate.
- Enhanced Supply Chain Reliability: The starting materials, specifically isatin derivatives and aromatic amines, are widely produced commodities with multiple global suppliers, reducing the risk of single-source dependency. This abundance ensures that lead times for raw material acquisition are short and predictable, allowing for just-in-time manufacturing strategies. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in reagent quality, further stabilizing the supply chain and reducing the frequency of batch failures or out-of-specification results.
- Scalability and Environmental Compliance: The method has been validated for expansion from gram-scale laboratory synthesis to potential industrial-scale application, indicating a smooth path for technology transfer. The use of DMF as a solvent, while requiring careful handling, is well-established in industrial settings with existing recovery infrastructure. Additionally, the avoidance of harsh acidic or basic workups typical of older methods simplifies wastewater treatment, aiding facilities in meeting stringent environmental compliance standards and reducing the overall ecological footprint of the manufacturing process.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this specific synthetic methodology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these nuances is essential for R&D teams evaluating this route for inclusion in their process development pipelines and for procurement teams assessing the long-term viability of the supply source.
Q: What are the primary advantages of this iron-catalyzed method over traditional trifluoroacetic anhydride routes?
A: This method utilizes inexpensive ferric chloride instead of costly precious metals and avoids the severe reaction conditions often associated with trifluoroacetic anhydride, resulting in better functional group tolerance and higher yields.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method can be expanded to the gram level and provides strong possibilities for industrial scale application due to the use of cheap, readily available raw materials and simple post-treatment procedures.
Q: What is the substrate scope for the R1 and R2 positions in this quinazolinone synthesis?
A: The method demonstrates wide applicability, tolerating various substituents including alkyl, halogen, methoxy, nitro, and aryl groups at both ortho-, meta-, and para-positions, allowing for the design of diverse molecular scaffolds.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the development of next-generation therapeutics. Our team of expert chemists has thoroughly analyzed the potential of this iron-catalyzed cyclization method and is prepared to leverage it for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from clinical trials to market launch is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone delivered meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to explore how this innovative synthesis can optimize your supply chain and reduce your overall development costs. Please contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities align with your strategic goals for high-purity pharmaceutical intermediates.