Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
Scalable Iron-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
The landscape of heterocyclic chemistry is constantly evolving, driven by the demand for more efficient and cost-effective pathways to bioactive scaffolds. A significant advancement in this domain is detailed in patent CN111675662A, which discloses a novel preparation method for 2-trifluoromethyl-substituted quinazolinone compounds. These nitrogen-containing fused-ring systems are pivotal in medicinal chemistry, serving as core structures for numerous drug candidates exhibiting anticancer, anticonvulsant, and anti-inflammatory properties. The introduction of a trifluoromethyl group into these frameworks is particularly strategic, as it enhances metabolic stability, lipophilicity, and bioavailability, making these molecules highly desirable for modern drug discovery programs. This patent presents a breakthrough by utilizing a tandem cyclization reaction catalyzed by inexpensive metallic iron, specifically ferric chloride, which stands in stark contrast to traditional methods that often rely on costly precious metals or harsh reaction conditions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinones bearing trifluoromethyl functionalities has been fraught with significant challenges that hinder their widespread adoption in large-scale manufacturing. Conventional literature reports predominantly describe methods utilizing trifluoromethyl-containing synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate reacting with substrates like anthranilamide or isatoic anhydride. These traditional pathways are frequently limited by severe reaction conditions that require stringent control of temperature and pressure, posing safety risks and operational complexities in a plant setting. Furthermore, the starting materials employed in these older methods are often expensive and less readily available, driving up the overall cost of goods. Perhaps most critically for process chemists, these legacy routes often suffer from narrow substrate scopes and mediocre yields, making it difficult to generate diverse libraries of analogs for structure-activity relationship (SAR) studies without incurring prohibitive costs and waste.
The Novel Approach
In a transformative shift, the methodology outlined in CN111675662A leverages readily available trifluoroethylimide chloride and isatin as starting materials to construct the quinazolinone core efficiently. This new approach bypasses the need for expensive activated trifluoroacetylating agents, instead relying on a robust iron-catalyzed system that operates under relatively mild conditions. The reaction demonstrates exceptional functional group tolerance, accommodating a wide array of substituents on both the aromatic amine and the isatin moieties, which is crucial for generating diverse chemical space. As illustrated in the structural diversity of the products below, this method allows for the precise installation of halogens, alkyl groups, and electron-withdrawing nitro groups without compromising the integrity of the final scaffold.
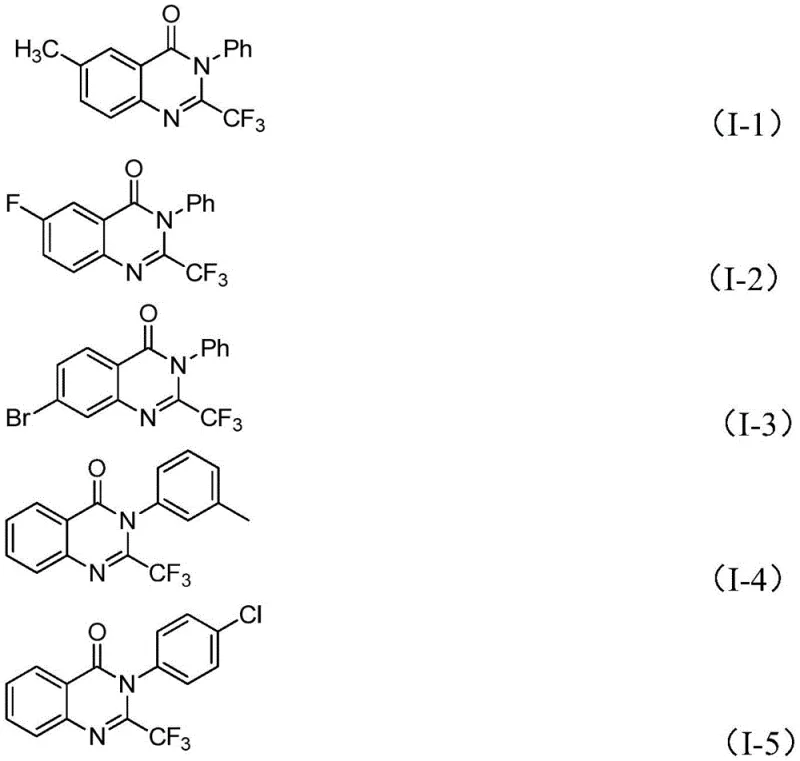
The versatility of this synthetic route is further evidenced by the successful preparation of various derivatives, including known compounds like I-2, I-4, and I-5, confirming the reliability of the process for producing established reference standards as well as novel entities. By shifting the paradigm from precious metal catalysis or harsh stoichiometric reagents to a base-metal catalyzed system, this invention offers a sustainable and economically viable pathway for the production of high-value pharmaceutical intermediates.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this innovation lies in the elegant mechanistic pathway facilitated by the ferric chloride catalyst. The reaction is proposed to proceed through a tandem sequence initiated by the base-promoted formation of an intermolecular carbon-nitrogen bond between the trifluoroethylimidoyl chloride and the isatin substrate. This initial step generates a trifluoroacetamidine intermediate, which then undergoes a critical iron-catalyzed decarbonylation cyclization. The presence of the iron catalyst is essential for activating the specific bonds required for ring closure while simultaneously facilitating the loss of the carbonyl group, a transformation that is often difficult to achieve with high selectivity using other metal systems. Following the cyclization, an isomerization step occurs to yield the thermodynamically stable 2-trifluoromethyl-substituted quinazolinone final product. This multi-step cascade occurring in a single pot exemplifies the efficiency of modern atom-economical synthesis.
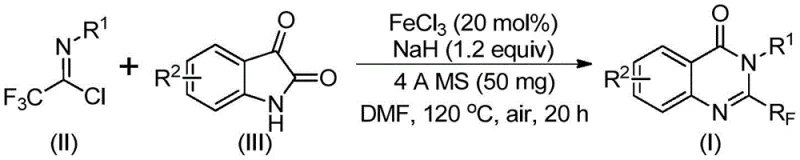
From an impurity control perspective, the use of 4A molecular sieves in the reaction mixture plays a pivotal role in maintaining high purity profiles. By sequestering trace amounts of water generated during the reaction or present in the solvent, the molecular sieves prevent the hydrolysis of the sensitive imidoyl chloride starting material and the intermediate species. This careful management of the reaction environment ensures that side reactions, such as the formation of carboxylic acid byproducts or incomplete cyclization, are minimized. Consequently, the crude reaction mixture typically exhibits a clean profile, simplifying the downstream purification process and ensuring that the final active pharmaceutical ingredient (API) intermediate meets stringent quality specifications required by regulatory bodies.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific operational parameters to maximize yield and safety. The process begins with the careful combination of the iron catalyst, sodium hydride base, and molecular sieves in an anhydrous aprotic solvent, with DMF being the preferred medium due to its superior solubilizing power for the polar intermediates. The reaction profile involves a two-stage heating protocol: an initial incubation at 40°C to allow for the gentle formation of the C-N bond, followed by a higher temperature phase at 120°C to drive the energetically demanding decarbonylation and cyclization steps to completion. Detailed standardized operating procedures for this transformation are provided below to ensure reproducibility and safety compliance.
- Combine ferric chloride catalyst, sodium hydride, 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin in an organic solvent such as DMF.
- Initiate the reaction at a mild temperature of 40°C for approximately 8 to 10 hours to facilitate initial bond formation.
- Increase the temperature to 120°C and maintain for 18 to 20 hours to complete the decarbonylation and cyclization, followed by purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this iron-catalyzed methodology represents a significant opportunity for cost optimization and risk mitigation. The replacement of expensive noble metal catalysts with commodity-grade ferric chloride drastically reduces the raw material cost per kilogram of product, directly impacting the bottom line. Moreover, the reliance on isatin and substituted anilines as starting materials leverages a supply chain that is well-established and robust, reducing the risk of shortages that often plague specialized fluorinated reagents. The operational simplicity of the process, which does not require exotic equipment or extreme cryogenic conditions, further contributes to substantial cost savings in terms of energy consumption and capital expenditure for reactor infrastructure.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts removes the need for expensive metal scavenging steps and complex recycling protocols, which are often mandatory when using palladium or rhodium systems. This simplification of the downstream processing workflow leads to a significant reduction in overall manufacturing costs, as the purification can be achieved through standard silica gel chromatography or crystallization without the burden of residual heavy metal limits. Additionally, the high atom economy of the tandem reaction minimizes waste generation, aligning with green chemistry principles and reducing waste disposal fees.
- Enhanced Supply Chain Reliability: The starting materials, specifically isatin and various substituted aromatic amines, are bulk chemicals produced by multiple global suppliers, ensuring a stable and competitive supply base. This diversification of sourcing options mitigates the risk of supply disruptions that can occur when relying on single-source specialty fluorinated building blocks. The robustness of the reaction conditions also means that the synthesis is less susceptible to minor fluctuations in raw material quality, providing greater consistency in production schedules and delivery timelines for downstream customers.
- Scalability and Environmental Compliance: The patent data confirms that the reaction has been successfully scaled to the gram level with consistent results, indicating a clear path toward kilogram and ton-scale commercial production. The use of DMF as a solvent, while requiring proper handling, is a standard industrial solvent with well-defined recovery and recycling protocols, facilitating compliance with environmental regulations. The absence of highly toxic or volatile reagents simplifies the safety profile of the plant, reducing the need for specialized containment systems and lowering insurance and operational safety costs.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis technology, derived directly from the experimental data and beneficial effects described in the patent documentation. These insights are intended to clarify the practical implications of adopting this method for the production of quinazolinone-based pharmaceutical intermediates. Understanding these nuances is critical for R&D teams evaluating process feasibility and procurement teams assessing long-term supply strategies.
Q: What are the primary advantages of this iron-catalyzed method over traditional synthesis?
A: This method utilizes inexpensive iron catalysts and readily available starting materials like isatin, avoiding the harsh conditions and expensive synthons like trifluoroacetic anhydride required by conventional routes, resulting in significantly higher yields and better functional group tolerance.
Q: What is the substrate scope for R1 and R2 groups in this reaction?
A: The process demonstrates excellent versatility, accommodating various substituted aryl groups for R1 (including methyl, fluoro, chloro, bromo, and nitro substituents) and diverse groups for R2 such as alkyl, halogen, or methoxy, allowing for the design of a wide library of derivatives.
Q: Is this synthesis protocol suitable for industrial scale-up?
A: Yes, the patent explicitly confirms that the methodology is operationally simple and has been successfully demonstrated at the gram level, indicating strong potential for commercial scale-up due to the use of stable reagents and standard organic solvents like DMF.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust and scalable synthetic routes in the development of next-generation therapeutics. Our team of expert process chemists has extensively evaluated the iron-catalyzed cyclization methodology described in CN111675662A and possesses the technical capability to implement this route effectively. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from benchtop discovery to full-scale manufacturing. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone intermediate meets the highest industry standards for potency and impurity profiles.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis can be tailored to your specific project needs. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this iron-catalyzed process for your specific volume requirements. We encourage you to contact us today to request specific COA data for our catalog compounds or to initiate a discussion on route feasibility assessments for your proprietary targets, ensuring a reliable supply of high-quality intermediates for your drug development pipeline.