Advanced Metal-Free Synthesis of Trifluoromethyl Triazoles for Commercial Scale-Up
Advanced Metal-Free Synthesis of Trifluoromethyl Triazoles for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust synthetic routes for nitrogen-containing heterocycles, particularly 1,2,4-triazoles, which serve as privileged scaffolds in numerous bioactive molecules. As highlighted in patent CN113105402B, a novel preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds has been developed that addresses critical bottlenecks in traditional manufacturing. This technology leverages a metal-free, iodine-promoted cascade reaction that operates under relatively mild conditions without the stringent requirement for anhydrous or oxygen-free environments. The significance of this chemical innovation is underscored by the prevalence of the triazole motif in blockbuster drugs such as Maraviroc, Sitagliptin, and Deferasirox, where the introduction of a trifluoromethyl group often enhances metabolic stability and lipophilicity. For R&D directors and process chemists, this patent represents a paradigm shift towards more sustainable and operationally simple protocols for generating high-value pharmaceutical intermediates.
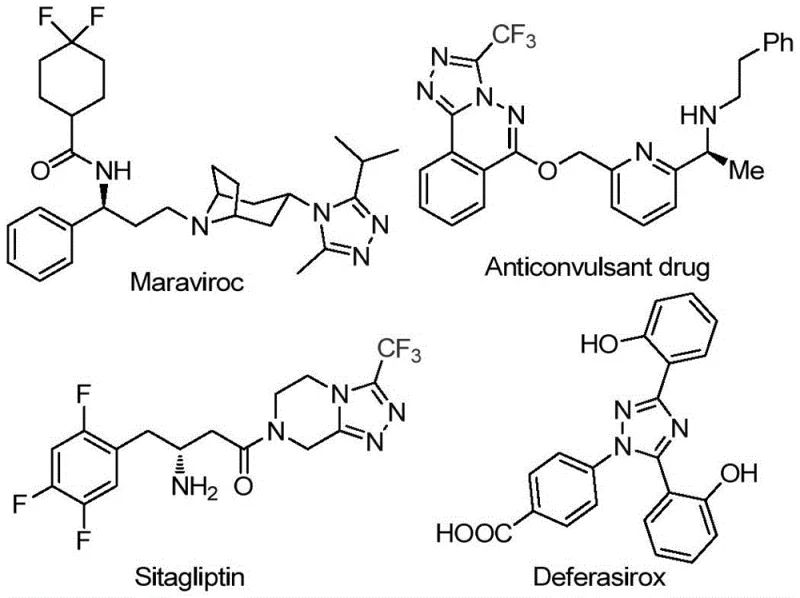
The structural versatility of these compounds allows for extensive modification at the 3, 4, and 5 positions, enabling medicinal chemists to fine-tune pharmacokinetic properties. By utilizing readily available aryl ethanones and trifluoroacetimidoyl hydrazides as starting materials, this method bypasses the need for complex, multi-step sequences often associated with triazole synthesis. The ability to introduce both acyl and trifluoromethyl groups simultaneously in a single pot is a distinct advantage, streamlining the supply chain for key API precursors. This report analyzes the technical merits of this invention, contrasting it with conventional methodologies and evaluating its potential for large-scale commercial adoption by global chemical manufacturers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles has relied heavily on transition metal catalysis or harsh reaction conditions that pose significant challenges for industrial scalability. Traditional routes often necessitate the use of expensive copper or palladium catalysts, which not only inflate raw material costs but also introduce severe complications regarding residual metal removal—a critical quality attribute for pharmaceutical ingredients. Furthermore, many existing protocols require strictly anhydrous and anaerobic conditions, demanding specialized equipment and inert gas manifolds that increase capital expenditure and operational complexity. The reliance on pre-functionalized starting materials, such as acid chlorides or hydrazonoyl halides, often involves hazardous reagents and generates substantial stoichiometric waste, conflicting with modern green chemistry principles. Additionally, the separation of byproducts in these metal-catalyzed reactions can be tedious, frequently requiring extensive chromatographic purification that limits throughput and increases production lead times.
The Novel Approach
In stark contrast, the methodology disclosed in CN113105402B employs a transition-metal-free strategy driven by elemental iodine and dimethyl sulfoxide (DMSO). This approach fundamentally simplifies the reaction setup by eliminating the need for gloveboxes or rigorous drying of solvents, thereby reducing the barrier to entry for manufacturing facilities. The core innovation lies in the tandem sequence where aryl ethanones undergo iodination and Kornblum oxidation in situ to form aryl diketones, which subsequently condense with trifluoroacetimidoyl hydrazides. This one-pot design minimizes unit operations and solvent consumption, directly translating to improved process mass intensity (PMI). The use of inexpensive inorganic bases like sodium dihydrogen phosphate and pyridine further optimizes the cost profile, making this route highly attractive for procurement teams focused on margin improvement. The reaction proceeds efficiently at temperatures between 110°C and 130°C, conditions that are easily achievable in standard stainless steel reactors without requiring cryogenic cooling or high-pressure vessels.
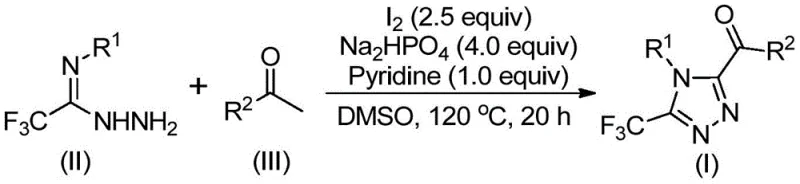
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of oxidation and cyclocondensation steps that ensures high regioselectivity and yield. Initially, the aryl ethanone reacts with molecular iodine in DMSO to undergo alpha-iodination followed by Kornblum oxidation, effectively converting the methyl ketone into an alpha-dicarbonyl species. This oxidative step is crucial as it generates the electrophilic 1,2-dicarbonyl intermediate required for ring closure. Subsequently, the nucleophilic attack by the amino group of the trifluoroacetimidoyl hydrazide on one of the carbonyl carbons leads to the formation of a hydrazone intermediate. Under the continued influence of iodine and the basic environment provided by pyridine and phosphate salts, this hydrazone undergoes intramolecular cyclization. The final aromatization step yields the stable 3,4,5-trisubstituted 1,2,4-triazole core. This mechanism avoids the formation of regioisomers often seen in non-directed cyclizations, ensuring a clean impurity profile that simplifies downstream processing.
From an impurity control perspective, the choice of reagents plays a pivotal role in minimizing side reactions. The use of sodium dihydrogen phosphate acts as a buffer, maintaining a pH environment that favors cyclization over hydrolysis of the sensitive imine bonds. The stoichiometry of iodine is carefully balanced; typically, 2.5 equivalents are used to drive the oxidation to completion without causing excessive halogenation of the aromatic rings. The substrate scope is remarkably broad, tolerating various electronic environments on both the N-aryl and C-acyl moieties. Electron-rich substrates bearing methoxy or methyl groups react efficiently, as do electron-deficient systems containing chloro or fluoro substituents. This electronic tolerance suggests that the rate-determining step is likely the initial oxidation or the cyclization itself, rather than the nucleophilic attack, allowing for consistent performance across a diverse library of analogues.
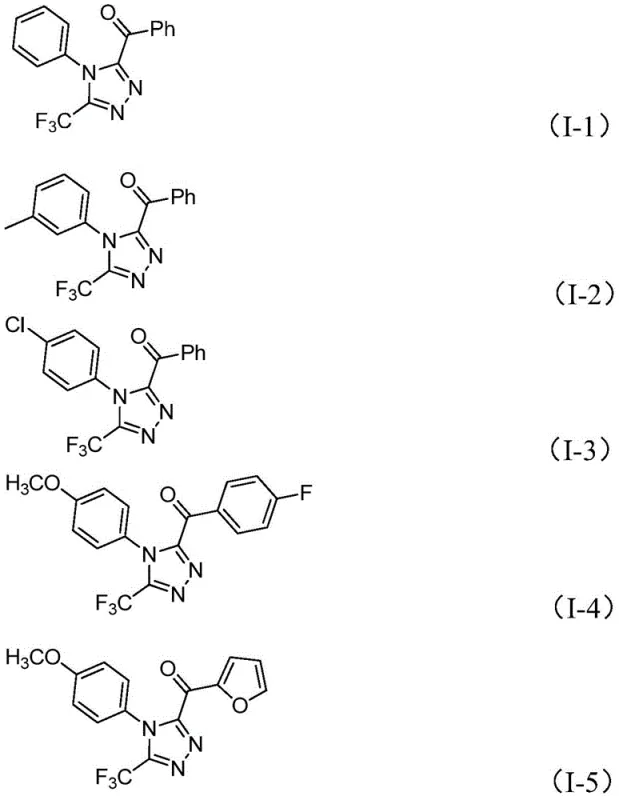
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The practical execution of this synthesis is designed for operational simplicity, making it accessible for both laboratory discovery and pilot plant operations. The process begins by dissolving the aryl ethanone and a portion of elemental iodine in DMSO, followed by heating to initiate the oxidation phase. Once the intermediate is formed, the remaining reagents including the hydrazide, base, and additional iodine are introduced directly into the same vessel. This telescoped approach eliminates the need to isolate the unstable diketone intermediate, reducing material loss and exposure to potentially hazardous substances. The reaction mixture is then heated to the final temperature range of 110°C to 130°C and maintained for 12 to 20 hours to ensure full conversion. Upon completion, the workup involves a straightforward filtration and silica gel treatment, followed by standard column chromatography to achieve high purity levels suitable for biological testing or further derivatization.
- Oxidation Phase: React aryl ethyl ketone with elemental iodine in DMSO at 90-110°C for 4-6 hours to generate the aryl diketone intermediate via Kornblum oxidation.
- Cyclization Phase: Add trifluoroethylimide hydrazide, additional iodine, sodium dihydrogen phosphate, and pyridine, then heat to 110-130°C for 12-20 hours.
- Purification: Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the target triazole compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis offers tangible strategic benefits beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the raw material portfolio; aryl ethanones and trifluoroacetimidoyl hydrazides are commodity chemicals available from multiple global suppliers, mitigating the risk of single-source dependency. By removing the requirement for precious metal catalysts, manufacturers can eliminate the costly and time-consuming steps associated with metal scavenging and validation, which are mandatory for API production. This reduction in processing steps directly correlates to a shorter overall cycle time, enabling faster response to market demands and reducing inventory holding costs. Furthermore, the robustness of the reaction against moisture and oxygen means that standard industrial reactors can be utilized without specialized modifications, lowering capital barriers for contract manufacturing organizations (CMOs) looking to offer this capability.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts such as palladium or copper significantly lowers the direct material cost per kilogram of product. Additionally, the avoidance of strict anhydrous conditions reduces energy consumption associated with solvent drying and inert gas purging. The high atom economy of the tandem reaction minimizes waste generation, leading to lower disposal costs and improved environmental compliance metrics. These cumulative efficiencies result in a more competitive cost structure for the final pharmaceutical intermediate, allowing for better margin management in volatile markets.
- Enhanced Supply Chain Reliability: Utilizing widely available starting materials ensures a stable supply chain that is less susceptible to geopolitical disruptions or shortages of specialized reagents. The simplicity of the reaction conditions allows for flexible manufacturing scheduling, as the process does not require dedicated lines for air-sensitive chemistry. This flexibility enhances the ability to scale production rapidly from gram to multi-kilogram quantities without significant process re-engineering. Consequently, lead times for delivering high-purity intermediates can be significantly reduced, supporting just-in-time manufacturing models for downstream API producers.
- Scalability and Environmental Compliance: The use of DMSO, a high-boiling polar aprotic solvent, facilitates heat transfer and mixing in large-scale reactors, ensuring consistent reaction performance upon scale-up. The absence of heavy metals simplifies the wastewater treatment process, as there is no need for complex metal precipitation or recovery systems. This aligns with increasingly stringent environmental regulations regarding heavy metal discharge, reducing the regulatory burden on manufacturing sites. The process generates minimal hazardous byproducts, contributing to a safer working environment and lower insurance premiums related to chemical handling risks.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a reliable basis for decision-making. Understanding these nuances is essential for R&D teams evaluating route selection and for procurement professionals assessing supplier capabilities. The answers reflect the balance between chemical efficiency and practical manufacturability that defines this innovative approach.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the method described in patent CN113105402B utilizes elemental iodine as a non-metal promoter, eliminating the need for costly palladium or copper catalysts and simplifying downstream purification.
Q: What is the role of DMSO in this reaction mechanism?
A: DMSO serves a dual purpose as both the solvent and the oxidant source. It participates in the Kornblum oxidation of the aryl ketone to an aryl diketone, which is a critical prerequisite for the subsequent cyclization.
Q: Can this method tolerate diverse functional groups on the aromatic rings?
A: Yes, the protocol demonstrates excellent functional group tolerance, successfully accommodating electron-donating groups like methoxy and methyl, as well as electron-withdrawing groups such as chloro and fluoro substituents.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic methodologies play in accelerating drug development timelines. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that laboratory successes are seamlessly translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee the quality of every batch. Our commitment to excellence extends beyond mere compliance; we actively collaborate with clients to optimize routes for cost and sustainability, leveraging technologies like the iodine-promoted triazole synthesis to deliver superior value.
We invite you to engage with our technical procurement team to discuss how this advanced manufacturing capability can support your pipeline. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits specific to your project volume. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing you to make informed decisions with confidence. Let us be your trusted partner in navigating the complexities of fine chemical synthesis and securing a reliable supply of high-quality pharmaceutical intermediates.