Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Intermediates
Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Intermediates
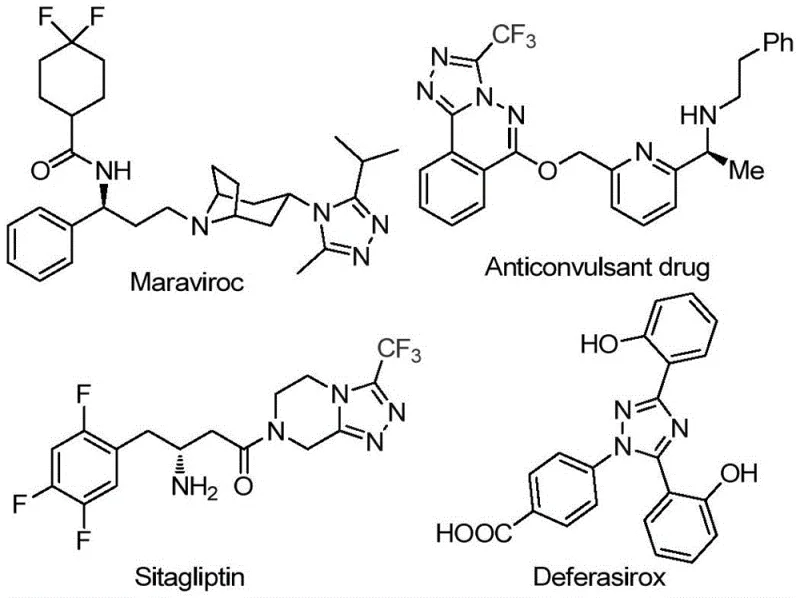
The pharmaceutical industry continuously seeks robust synthetic routes for nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which enhance metabolic stability and bioavailability. Patent CN113105402A discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds, a structural core prevalent in blockbuster drugs such as Maraviroc and Sitagliptin. This technology leverages a non-metallic iodine-promoted oxidative cyclization strategy, utilizing dimethyl sulfoxide (DMSO) as both solvent and oxidant source. By circumventing the need for expensive transition metal catalysts and严苛 anhydrous conditions, this innovation addresses critical pain points in cost reduction in pharmaceutical intermediate manufacturing. The methodology demonstrates exceptional substrate tolerance, accommodating various aryl and heteroaryl groups, thereby positioning itself as a versatile platform for generating high-purity scaffolds essential for modern drug discovery pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic pathways for constructing polysubstituted 1,2,4-triazole rings often rely heavily on transition metal catalysis, such as copper or palladium-mediated coupling reactions, which introduce significant economic and environmental burdens. These conventional protocols frequently necessitate rigorous exclusion of moisture and oxygen, requiring specialized equipment and inert gas atmospheres that drastically increase operational complexity and capital expenditure. Furthermore, the removal of trace heavy metal residues from the final active pharmaceutical ingredient (API) is a stringent regulatory requirement, often demanding additional purification steps like scavenging or recrystallization that erode overall yield. The reliance on pre-functionalized starting materials, such as acid chlorides or hydrazonoyl halides, further complicates the supply chain, as these reagents can be unstable, hazardous, or cost-prohibitive for large-scale commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
The disclosed invention revolutionizes this landscape by employing a tandem oxidative cyclization driven by elemental iodine and DMSO, effectively merging the oxidation of arylethanones and the subsequent heterocycle formation into a streamlined one-pot sequence. This approach initiates with the iodine-mediated Kornblum oxidation of readily available arylethanones to generate reactive alpha-diketone intermediates in situ, which then undergo condensation with trifluoroacetimidoyl hydrazides. The absence of toxic heavy metals not only aligns with green chemistry principles but also simplifies the downstream processing, allowing for straightforward filtration and chromatographic purification. By operating under aerobic conditions with cheap, shelf-stable reagents, this method significantly lowers the barrier to entry for producing trifluoromethylated triazoles, offering a reliable alternative for reliable pharmaceutical intermediate suppliers aiming to optimize their production portfolios.
Mechanistic Insights into Iodine-Mediated Oxidative Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of oxidation and condensation events orchestrated by the iodine-DMSO system. Initially, elemental iodine activates the methyl group of the arylethanone substrate, facilitating its oxidation by DMSO to form an alpha-dicarbonyl species, a critical electrophilic intermediate. This step mimics the classic Kornblum oxidation but is uniquely adapted here to function in the presence of nitrogen nucleophiles. Subsequently, the trifluoroacetimidoyl hydrazide acts as a bidentate nucleophile, attacking the carbonyl centers to form a hydrazone linkage. The presence of sodium dihydrogen phosphate and pyridine serves to buffer the acidic byproducts (HI) generated during the oxidation, maintaining the optimal pH for the subsequent cyclization step.
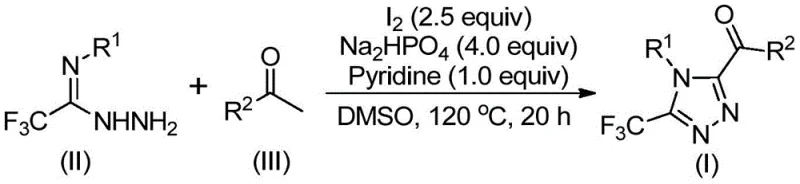
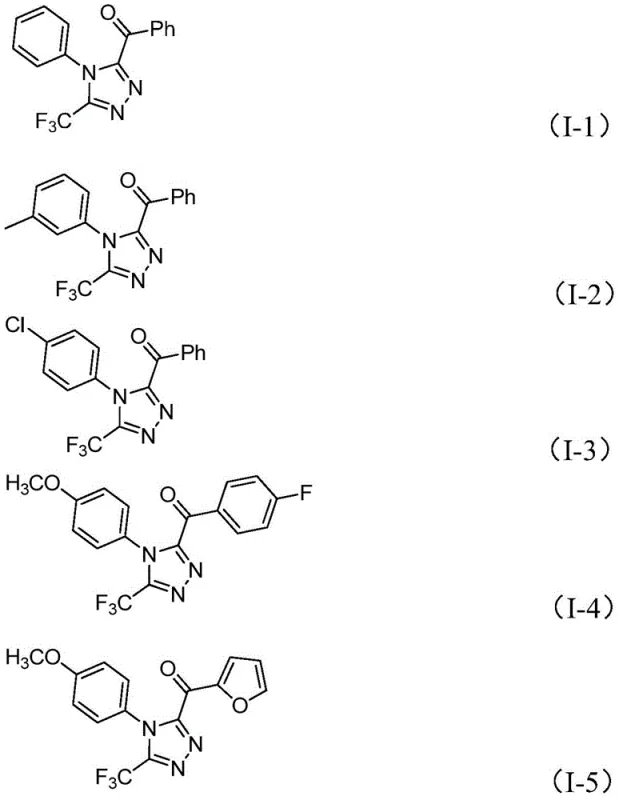
Following the condensation, the reaction mixture undergoes an intramolecular cyclization promoted by the continued presence of iodine and base, closing the five-membered triazole ring while expelling water. The introduction of the trifluoromethyl group at the 3-position is inherent to the hydrazide starting material, ensuring precise regiocontrol without the need for post-synthetic fluorination, which is often dangerous and low-yielding. This mechanistic elegance ensures that side reactions, such as over-oxidation or polymerization, are minimized, leading to cleaner reaction profiles. The robustness of this mechanism against varying electronic properties of the aryl substituents (electron-donating methoxy or electron-withdrawing chloro groups) underscores its utility for synthesizing diverse libraries of high-purity pharmaceutical intermediates with consistent quality.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The operational simplicity of this protocol makes it highly attractive for process chemists looking to implement rapid synthesis campaigns. The procedure involves a sequential addition strategy where the arylethanone and iodine are first heated in DMSO to generate the oxidized intermediate, followed by the addition of the hydrazide and base components for the cyclization phase. This two-stage temperature control (initially 90-110°C, then 110-130°C) is crucial for maximizing conversion while minimizing decomposition. Detailed standardized synthesis steps, including precise molar ratios and workup procedures, are provided below to ensure reproducibility across different laboratory settings.
- Mix arylethanone and elemental iodine in DMSO, heating to 90-110°C for 4-6 hours to generate the alpha-diketone intermediate via Kornblum oxidation.
- Add trifluoroacetimidoyl hydrazide, sodium dihydrogen phosphate, pyridine, and additional iodine to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours to facilitate dehydration condensation and intramolecular cyclization, followed by filtration and chromatographic purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this technology offers compelling advantages by decoupling production from the volatile supply chains associated with precious metal catalysts. The primary reagents—arylethanones, elemental iodine, and DMSO—are commodity chemicals available in bulk quantities from multiple global vendors, ensuring supply continuity and price stability. The elimination of noble metals like palladium removes a significant cost driver and mitigates the risk of supply disruptions caused by geopolitical factors affecting mining regions. Furthermore, the ability to run the reaction without strict inert atmosphere protection reduces the consumption of nitrogen or argon gases and lowers the energy footprint associated with maintaining glovebox or Schlenk line conditions.
- Cost Reduction in Manufacturing: The substitution of expensive transition metal catalysts with inexpensive elemental iodine results in a drastic reduction in raw material costs per kilogram of product. Additionally, the simplified purification process, which avoids complex metal scavenging resins, reduces the consumption of auxiliary materials and solvent volumes. The high atom economy of the tandem reaction means less waste generation, directly translating to lower waste disposal fees and a more sustainable manufacturing profile that aligns with corporate ESG goals.
- Enhanced Supply Chain Reliability: Since the starting materials are stable, non-hazardous solids or liquids with long shelf lives, inventory management becomes significantly more predictable compared to handling moisture-sensitive organometallic reagents. The robustness of the reaction conditions allows for flexible scheduling, as the process is not critically dependent on immediate anhydrous setups, reducing the lead time for batch preparation. This reliability ensures that reducing lead time for high-purity pharmaceutical intermediates is achievable, allowing manufacturers to respond swiftly to market demands.
- Scalability and Environmental Compliance: The use of DMSO, a high-boiling polar aprotic solvent, facilitates heat transfer and solubility of polar intermediates, making the transition from gram-scale optimization to multi-kilogram production seamless. The absence of heavy metal contaminants simplifies the environmental compliance dossier, as effluent streams do not require specialized treatment for metal removal. This ease of scale-up, combined with the use of benign reagents, positions this method as an ideal candidate for continuous flow processing or large-batch reactor campaigns in GMP facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this iodine-mediated synthesis. Understanding these nuances is vital for R&D teams evaluating this route for their specific pipeline candidates. The answers are derived directly from the experimental data and scope limitations outlined in the patent documentation, providing a realistic view of the technology's capabilities.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the patented method utilizes elemental iodine as a non-metallic promoter, eliminating the need for costly palladium or copper catalysts and simplifying downstream purification.
Q: What are the typical reaction conditions for this triazole formation?
A: The reaction proceeds in dimethyl sulfoxide (DMSO) under aerobic conditions, involving a sequential heating protocol starting at 90-110°C followed by 110-130°C, without requiring strict anhydrous environments.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the process uses cheap, commercially available raw materials like arylethanones and avoids sensitive conditions, making it highly amenable to scale-up from gram to multi-kilogram levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of efficient heterocycle synthesis in accelerating drug development timelines. Our technical team has extensively analyzed this iodine-promoted pathway and possesses the expertise to adapt it for custom synthesis projects requiring specific substitution patterns on the triazole core. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need grams for screening or tons for commercial launch, our capacity meets your needs. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of trifluoromethyl triazole intermediate meets the highest international standards.
We invite potential partners to leverage our technical acumen to optimize their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us to obtain specific COA data for similar structures and discuss route feasibility assessments for your target molecules, ensuring a seamless transition from bench-scale discovery to commercial reality.