Scalable Metal-Free Synthesis of 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazoles for Advanced Drug Discovery
Scalable Metal-Free Synthesis of 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazoles for Advanced Drug Discovery
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable, and cost-effective methodologies for constructing complex heterocyclic scaffolds that serve as the backbone of modern therapeutics. A significant breakthrough in this domain is detailed in Chinese Patent CN113307790B, which discloses a highly efficient preparation method for 3-quinolyl-5-trifluoromethyl substituted 1,2,4-triazole compounds. These specific heterocyclic structures are of immense value due to their prevalence in bioactive molecular frameworks, ranging from potent drug intermediates to functional materials used in organic light-emitting diodes (OLEDs). The innovation lies in its ability to bypass traditional multi-step syntheses, offering a streamlined, one-pot oxidative cyclization strategy that significantly enhances the feasibility of producing these high-value intermediates for global supply chains.
For R&D directors and process chemists, the introduction of this technology represents a pivotal shift away from cumbersome synthetic routes that often plague early-stage drug discovery. The patent highlights a novel approach utilizing cheap and easily obtainable starting materials, specifically 2-methylquinolines and trifluoroacetimidohydrazides, coupled with an organocatalytic system. This eliminates the dependency on precious metal catalysts, thereby reducing the environmental footprint and simplifying the purification process. By leveraging this methodology, manufacturers can achieve high-purity pharmaceutical intermediates with improved impurity profiles, directly addressing the stringent quality control requirements of regulatory bodies worldwide.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinolyl-substituted 1,2,4-triazoles has been fraught with significant technical and economic challenges that hinder large-scale application. Traditional protocols often rely on quinoline-2-carboxylic acid as a primary raw material, necessitating a laborious five-step reaction sequence to arrive at the target molecule. This multi-step approach not only results in a dismal overall yield of approximately 17% but also requires severe reaction conditions that demand rigorous control over temperature and pressure. Furthermore, the accumulation of byproducts across multiple steps complicates the purification process, leading to higher solvent consumption and increased waste generation. For procurement managers, these inefficiencies translate into inflated costs of goods sold (COGS) and extended lead times, making the conventional route economically unviable for commercial manufacturing.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN113307790B introduces a direct oxidative cyclization pathway that dramatically simplifies the synthetic landscape. By employing tetrabutylammonium iodide (TBAI) and tert-butyl hydroperoxide (TBHP) as promoters, the reaction effectively converts 2-methylquinolines and trifluoroacetimidohydrazides into the desired triazole products in a single operational step. This novel approach operates under relatively mild conditions, typically between 80°C and 100°C, and does not require strict anhydrous or oxygen-free environments, which greatly reduces operational complexity. The versatility of this method allows for the introduction of diverse substituents on both the quinoline and the aryl rings, enabling the rapid generation of compound libraries for structure-activity relationship (SAR) studies without the bottleneck of complex synthesis.
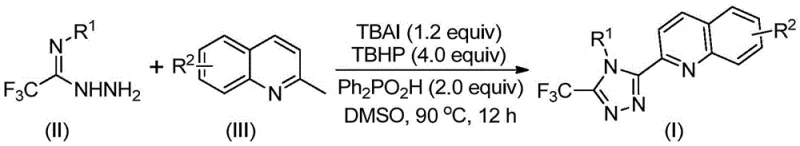
The strategic advantage of this new route is further amplified by its compatibility with a wide range of functional groups, ensuring that sensitive moieties remain intact during the transformation. This robustness is critical for the synthesis of complex drug candidates where specific substitution patterns are required for biological activity. By collapsing a five-step sequence into a single pot, the novel approach not only boosts the overall yield significantly—often exceeding 70% for various substrates—but also minimizes the exposure of intermediates to potential degradation. For supply chain heads, this translates to a more reliable production schedule and a substantial reduction in the inventory of hazardous intermediates, aligning perfectly with modern green chemistry principles and safety standards.
Mechanistic Insights into TBAI/TBHP Catalyzed Oxidative Cyclization
Understanding the mechanistic underpinnings of this transformation is essential for R&D teams aiming to optimize the process for specific substrates or scale-up scenarios. The reaction is believed to proceed through a radical-mediated pathway initiated by the interaction between tetrabutylammonium iodide and tert-butyl hydroperoxide. Initially, the TBAI acts as a catalyst to activate the TBHP, generating reactive iodine species that facilitate the oxidation of the methyl group on the 2-methylquinoline substrate. This oxidation step converts the methyl group into an aldehyde functionality in situ, forming a 2-quinolinecarbaldehyde intermediate without the need for isolation. This transient aldehyde then undergoes a condensation reaction with the trifluoroacetimidohydrazide to form a dehydrated hydrazone intermediate, setting the stage for the subsequent cyclization event.
Following the formation of the hydrazone, the reaction proceeds through an oxidative iodination and intramolecular electrophilic substitution sequence. The iodine species generated in the catalytic cycle attack the hydrazone nitrogen, promoting the closure of the triazole ring. Subsequent aromatization steps finalize the formation of the stable 3-quinolyl-5-trifluoromethyl-1,2,4-triazole core. The presence of diphenylphosphoric acid as an additive plays a crucial role in stabilizing the transition states and enhancing the reaction efficiency, likely by acting as a proton shuttle or coordinating with the iodine species. This intricate interplay of reagents ensures high conversion rates and minimizes the formation of side products, resulting in a clean reaction profile that is highly desirable for industrial applications.
How to Synthesize 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazole Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and reaction parameters to maximize yield and purity. The protocol involves mixing the key components—tetrabutylammonium iodide, tert-butyl hydroperoxide aqueous solution, diphenylphosphoric acid, trifluoroacetimidohydrazide, and 2-methylquinoline—in a suitable organic solvent such as dimethyl sulfoxide (DMSO). The choice of solvent is critical, as DMSO has been shown to provide superior solubility for the starting materials and effectively promote the oxidative cyclization compared to other aprotic solvents. The reaction mixture is then heated to a temperature range of 80°C to 100°C and maintained under stirring for a period of 8 to 14 hours, ensuring complete consumption of the starting materials.
- Combine tetrabutylammonium iodide (TBAI), tert-butyl hydroperoxide (TBHP), diphenylphosphoric acid, trifluoroacetimidohydrazide, and 2-methylquinoline in an organic solvent such as DMSO.
- Heat the reaction mixture to a temperature range of 80°C to 100°C and maintain stirring for a duration of 8 to 14 hours to ensure complete conversion.
- Upon completion, filter the mixture, adsorb onto silica gel, and purify via column chromatography to isolate the target 3-quinolyl-5-trifluoromethyl-1,2,4-triazole product.
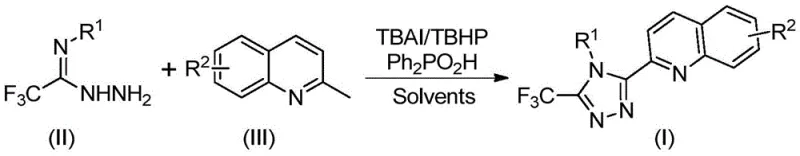
Upon completion of the reaction, the workup procedure is straightforward and designed for ease of operation. The reaction mixture is filtered to remove any insoluble particulates, and the filtrate is mixed with silica gel to facilitate purification. The final product is isolated via column chromatography, a standard technique in organic synthesis that allows for the separation of the target triazole from minor byproducts and unreacted starting materials. For detailed standard operating procedures and specific stoichiometric ratios tailored to different substrates, please refer to the comprehensive guide below.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this metal-free oxidative cyclization technology offers profound benefits for procurement strategies and supply chain resilience. The elimination of heavy metal catalysts is a primary driver for cost reduction, as it removes the necessity for expensive scavenging resins and complex purification steps required to meet residual metal limits in pharmaceutical ingredients. This simplification of the downstream processing significantly lowers the operational expenditure associated with manufacturing, making the final API intermediate more price-competitive in the global market. Furthermore, the use of commodity chemicals like TBAI and TBHP, which are widely available and inexpensive, ensures a stable supply of raw materials不受 geopolitical fluctuations that often affect rare earth or precious metal catalysts.
- Cost Reduction in Manufacturing: The streamlined one-pot nature of this synthesis drastically reduces the number of unit operations required, leading to significant savings in labor, energy, and solvent usage. By avoiding the multi-step sequence of traditional methods, manufacturers can reduce the overall production time and minimize the loss of material at each transfer stage. The high atom economy and excellent yields reported in the patent data suggest that the cost per kilogram of the final product can be substantially lowered, providing a clear margin advantage for generic drug manufacturers and custom synthesis providers alike.
- Enhanced Supply Chain Reliability: The reliance on readily available, non-hazardous starting materials enhances the robustness of the supply chain. Unlike specialized reagents that may have long lead times or single-source suppliers, the key components for this reaction are standard industrial chemicals with established global distribution networks. This accessibility mitigates the risk of production delays caused by raw material shortages, ensuring consistent delivery schedules for downstream clients. Additionally, the tolerance of the reaction to ambient conditions reduces the need for specialized infrastructure, allowing for flexible manufacturing across different facilities.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work efficiently from gram-scale experiments to larger batch sizes without significant loss in performance. The absence of toxic heavy metals simplifies waste treatment protocols, reducing the environmental burden and compliance costs associated with hazardous waste disposal. This alignment with green chemistry principles not only improves the corporate sustainability profile but also future-proofs the manufacturing process against increasingly stringent environmental regulations, ensuring long-term operational viability.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific projects, we have compiled a set of frequently asked questions based on the detailed technical disclosures within the patent literature. These answers address common concerns regarding catalyst toxicity, substrate flexibility, and the practical aspects of scaling the reaction for commercial production. Understanding these nuances is critical for making informed decisions about process adoption and integration into existing manufacturing workflows.
Q: Does this synthesis method require toxic heavy metal catalysts?
A: No, the method described in patent CN113307790B utilizes an organocatalytic system based on tetrabutylammonium iodide (TBAI) and tert-butyl hydroperoxide (TBHP), completely eliminating the need for expensive and toxic transition metal catalysts.
Q: What is the substrate scope for the quinoline component?
A: The process demonstrates excellent tolerance for various substituents on the quinoline ring, including hydrogen, methyl, methoxy, halogens (Cl, Br), and nitro groups at different positions, allowing for diverse structural modifications.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the reaction operates under relatively mild conditions (80-100°C) without strict anhydrous or oxygen-free requirements, uses commercially available reagents, and has been demonstrated to be easily expandable from gram-scale to larger production batches.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic methodologies like the one described in patent CN113307790B for accelerating drug development pipelines. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive high-quality intermediates consistently. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 3-quinolyl-5-trifluoromethyl-1,2,4-triazole delivered meets the highest industry standards for safety and efficacy.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this cutting-edge technology for their next-generation therapeutics. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in metal-free catalysis can drive efficiency and innovation in your supply chain.