Scalable Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Advanced Drug Discovery
Scalable Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazines for Advanced Drug Discovery
The rapid evolution of medicinal chemistry demands efficient access to privileged scaffolds that can modulate biological targets with high precision. Among these, trifluoromethyl-substituted 1,2,4-triazine compounds have emerged as critical structural motifs in the development of next-generation therapeutics, exhibiting potent anticancer, antifungal, and anti-inflammatory activities. As illustrated in the diverse bioactive molecules shown below, these heterocycles serve as key building blocks for complex pharmaceutical agents, including PI3Kα inhibitors and dual c-Met/VEGFR-2 inhibitors.
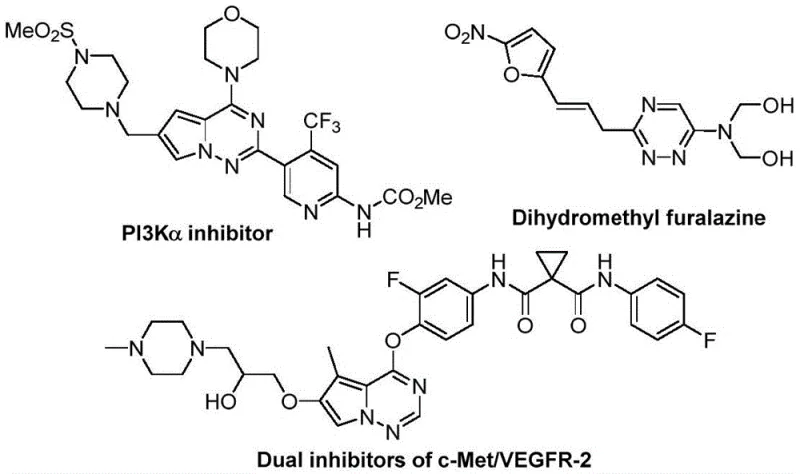
However, the incorporation of the trifluoromethyl group into the triazine core has historically presented significant synthetic challenges, often requiring harsh conditions or multi-step sequences. The recent disclosure in patent CN116253692A introduces a transformative preparation method that addresses these bottlenecks by utilizing a direct cycloaddition strategy. This innovation not only streamlines the synthetic route but also aligns perfectly with the green chemistry principles increasingly demanded by global regulatory bodies and procurement teams seeking sustainable supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the construction of the 1,2,4-triazine skeleton has relied heavily on the condensation of amidrazones with 1,2-diketones or alkynes, as well as multicomponent reactions involving hydrazides and dicarbonyl compounds. While these classical approaches are well-documented, they suffer from inherent inefficiencies that hinder their utility in modern high-throughput drug discovery and commercial manufacturing. A primary drawback is the frequent necessity for pre-functionalized substrates that are themselves difficult and costly to synthesize, thereby elongating the overall production timeline and inflating the cost of goods.
Furthermore, many conventional cyclization protocols demand the use of transition metal catalysts or strong acidic/basic conditions that are incompatible with sensitive functional groups often found in advanced intermediates. The requirement for rigorous exclusion of moisture and oxygen adds another layer of operational complexity, necessitating specialized equipment and increasing the risk of batch failure. Additionally, the structural diversity achievable through these older methods is often limited, restricting the chemical space available for medicinal chemists to explore structure-activity relationships effectively.
The Novel Approach
In stark contrast to these legacy techniques, the methodology described in CN116253692A leverages a synergistic [3+3] cycloaddition between readily available chlorohydrazones and trifluoroacetyl sulfur ylides. This novel pathway operates under remarkably mild conditions, specifically at temperatures between 20°C and 40°C, and crucially, proceeds efficiently in an ambient air atmosphere without the need for inert gas protection. The use of potassium carbonate as a benign, inexpensive inorganic base replaces costly and toxic organometallic catalysts, fundamentally altering the economic and safety profile of the synthesis.
This approach dramatically simplifies the post-reaction workup, as the absence of heavy metals eliminates the need for complex scavenging steps or extensive purification to meet strict residual metal specifications required for pharmaceutical ingredients. The reaction demonstrates exceptional tolerance for a wide array of substituents, including halogens, alkoxy groups, and trifluoromethyl moieties, allowing for the rapid generation of diverse libraries of trifluoromethyl-substituted 1,2,4-triazines. This flexibility empowers R&D teams to accelerate lead optimization campaigns while providing procurement managers with a robust, cost-effective route for scale-up.
Mechanistic Insights into Potassium Carbonate-Promoted [3+3] Cycloaddition
The mechanistic elegance of this transformation lies in the in situ generation of a reactive nitrile imine intermediate followed by a concerted cycloaddition event. As depicted in the general reaction scheme below, the process initiates with the deprotonation of the chlorohydrazone by potassium carbonate, leading to the elimination of hydrogen chloride and the formation of the electrophilic nitrile imine species. This highly reactive intermediate then engages the trifluoroacetyl sulfur ylide in a formal [3+3] cycloaddition, constructing the six-membered triazine ring with high regioselectivity.
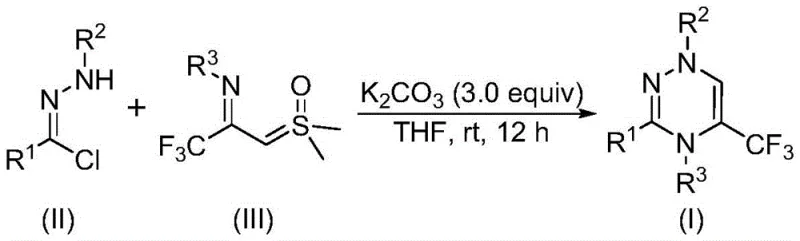
Following the ring closure, the elimination of dimethyl sulfoxide drives the reaction forward to yield the final aromatic triazine product. An alternative stepwise pathway involving intermolecular nucleophilic addition followed by intramolecular substitution is also plausible, yet both pathways converge to the same thermodynamically stable product. The choice of tetrahydrofuran (THF) as the solvent is particularly advantageous, as it effectively solubilizes both the organic substrates and the inorganic base, facilitating efficient mass transfer and ensuring consistent reaction kinetics across different scales.
From an impurity control perspective, the clean nature of this reaction minimizes the formation of side products commonly associated with radical pathways or metal-catalyzed couplings. The primary byproduct, dimethyl sulfoxide, is highly polar and water-soluble, making its removal during the aqueous workup straightforward and efficient. This high level of chemoselectivity ensures that the crude product profile is clean, reducing the burden on downstream purification units and maximizing the overall recovery of high-purity material suitable for subsequent coupling reactions in API synthesis.
How to Synthesize Trifluoromethyl Substituted 1,2,4-Triazine Efficiently
To implement this synthesis in a laboratory or pilot plant setting, operators should adhere to the optimized parameters established in the patent examples, which demonstrate reproducibility across fifteen distinct derivatives. The protocol involves charging a reactor with the chlorohydrazone, the trifluoroacetyl sulfur ylide, and three equivalents of potassium carbonate in THF, followed by stirring at room temperature for approximately 12 hours. Detailed standardized operating procedures regarding stoichiometry, specific substrate variations, and isolation techniques are provided in the technical guide below.
- Prepare the reaction mixture by adding potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide into an organic solvent such as tetrahydrofuran.
- Stir the mixture at room temperature (20-40°C) in an air atmosphere for 10 to 14 hours to allow the [3+3] cycloaddition to proceed.
- Upon completion, filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the target trifluoromethyl substituted 1,2,4-triazine compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement specialists and supply chain directors, the adoption of this metal-free synthesis route offers compelling strategic advantages that directly impact the bottom line and operational resilience. By shifting away from precious metal catalysts, manufacturers can insulate their production costs from the volatile pricing of commodities like palladium or rhodium, ensuring more predictable budgeting for long-term projects. Furthermore, the simplified regulatory landscape regarding heavy metal residues reduces the analytical testing burden and accelerates the release of batches for clinical or commercial use.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and ligands results in a drastic reduction in raw material costs, while the use of commodity chemicals like potassium carbonate further enhances economic efficiency. The mild reaction conditions negate the need for energy-intensive heating or cooling systems, leading to substantial savings in utility consumption over the lifecycle of the product. Additionally, the streamlined workup process reduces solvent usage and labor hours associated with complex purification steps, contributing to a leaner and more cost-competitive manufacturing process.
- Enhanced Supply Chain Reliability: The starting materials, including chlorohydrazones and sulfur ylides, are derived from widely available bulk chemicals, mitigating the risk of supply disruptions often associated with specialized reagents. The robustness of the reaction in air means that production is not contingent on the availability of high-purity nitrogen or argon, simplifying facility requirements and allowing for greater flexibility in manufacturing locations. This accessibility ensures a continuous and reliable flow of high-purity pharmaceutical intermediates, safeguarding against delays in downstream API production schedules.
- Scalability and Environmental Compliance: The absence of toxic heavy metals significantly simplifies waste management and disposal, aligning with stringent environmental regulations and corporate sustainability goals. The process is inherently scalable, as demonstrated by the successful synthesis of gram-scale quantities without loss of efficiency, paving the way for seamless technology transfer to multi-kilogram or ton-scale production. This scalability, combined with the use of greener reagents, positions this method as a future-proof solution for the sustainable manufacturing of complex heterocyclic intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel trifluoromethyl triazine synthesis platform. These insights are derived directly from the experimental data and comparative analysis presented in the patent literature, providing clarity for technical decision-makers evaluating this technology for integration into their existing pipelines.
Q: What are the primary advantages of this synthesis method over traditional routes?
A: This method eliminates the need for expensive and toxic heavy metal catalysts, operates at room temperature in air, and uses inexpensive potassium carbonate as a promoter, significantly simplifying purification and reducing operational costs.
Q: What is the typical yield range for these trifluoromethyl triazine derivatives?
A: According to the patent data, the reaction demonstrates robust efficiency with isolated yields ranging from 62% to 87% across various substrates, indicating high reliability for diverse functional groups.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is highly scalable as it avoids sensitive reagents requiring inert atmospheres, utilizes common solvents like THF, and employs simple workup procedures compatible with industrial equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl 1,2,4-Triazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality heterocyclic intermediates play in accelerating drug discovery and development timelines. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from bench-scale innovation to industrial reality is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of trifluoromethyl substituted 1,2,4-triazine meets the exacting standards required by global pharmaceutical partners.
We invite you to collaborate with us to leverage this advanced synthesis technology for your specific project needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities can optimize your supply chain and reduce your overall cost of goods.