Advanced Synthesis of Trifluoromethyl 1,2,4-Triazines for High-Purity Pharmaceutical Intermediates
Advanced Synthesis of Trifluoromethyl 1,2,4-Triazines for High-Purity Pharmaceutical Intermediates
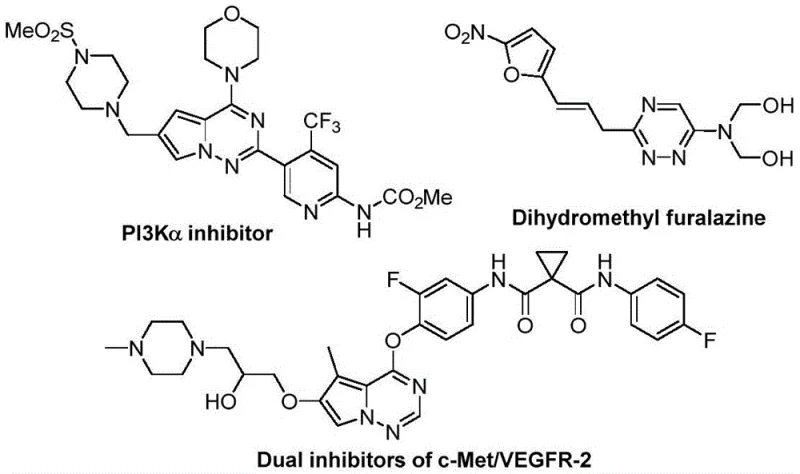
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting potent biological activities. Patent CN116253692A introduces a groundbreaking preparation method for trifluoromethyl-substituted 1,2,4-triazine compounds, addressing critical bottlenecks in the production of high-value pharmaceutical intermediates. These heterocyclic structures are pivotal in the design of anticancer, antifungal, and anti-inflammatory agents, as evidenced by their presence in complex drug candidates such as PI3Kα inhibitors and dual c-Met/VEGFR-2 inhibitors shown in recent literature. The strategic incorporation of the trifluoromethyl group further enhances metabolic stability and lipophilicity, making these compounds highly desirable for drug discovery programs. This new methodology offers a streamlined, cost-effective alternative to traditional synthesis, positioning it as a vital technology for reliable pharmaceutical intermediate suppliers aiming to optimize their manufacturing portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the 1,2,4-triazine core has relied heavily on the condensation reaction of amidrazones with 1,2-diketones or alkynes, as well as multicomponent reactions involving hydrazides and dicarbonyl compounds. While effective in specific contexts, these conventional pathways often suffer from significant drawbacks that hinder their industrial applicability. Traditional methods frequently necessitate the pre-synthesis of complex reaction substrates, leading to increased step counts and lower overall atom economy. Furthermore, many established protocols require harsh reaction conditions, including elevated temperatures or the use of expensive transition metal catalysts, which introduce challenges in impurity control and downstream purification. The structural diversity achievable through these older routes is often limited, restricting the ability of R&D teams to rapidly explore structure-activity relationships (SAR) around the triazine scaffold. Consequently, there is a pressing demand for more robust and versatile synthetic strategies that can overcome these efficiency barriers.
The Novel Approach
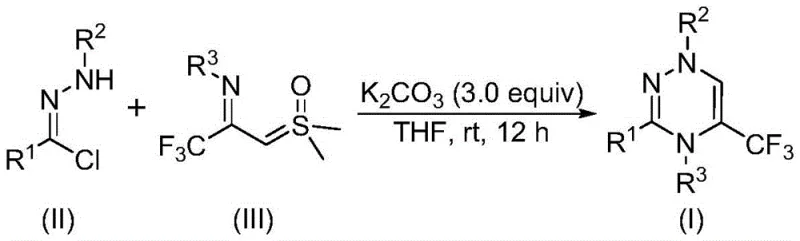
The invention disclosed in CN116253692A presents a paradigm shift by utilizing a direct [3+3] cycloaddition strategy between chlorohydrazones and trifluoroacetyl sulfur ylides. This novel approach eliminates the need for precious metal catalysts, relying instead on inexpensive and non-toxic potassium carbonate as a promoter. The reaction proceeds smoothly at room temperature (20-40°C) under an air atmosphere, removing the stringent requirement for inert gas protection that typically inflates operational costs in fine chemical manufacturing. By employing readily available starting materials such as acyl chlorides and hydrazines, the method drastically simplifies the supply chain logistics. The general reaction scheme illustrates the elegant transformation where a nitrile imine intermediate, generated in situ, reacts synergistically with the sulfur ylide to construct the triazine ring with the elimination of dimethyl sulfoxide. This metal-free, ambient condition process represents a significant advancement in cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Metal-Free [3+3] Cycloaddition
The mechanistic pathway of this transformation is both elegant and efficient, driven by the unique reactivity of the sulfur ylide species. Under the promotion of potassium carbonate, the chlorohydrazone undergoes dehydrohalogenation to generate a highly reactive nitrile imine intermediate. This 1,3-dipole then engages in a concerted [3+3] cycloaddition with the trifluoroacetyl sulfur ylide, which acts as a three-carbon synthon equivalent. The reaction is characterized by the simultaneous formation of two new carbon-nitrogen bonds and one carbon-carbon bond, effectively closing the six-membered triazine ring in a single operational step. The expulsion of dimethyl sulfoxide (DMSO) serves as the thermodynamic driving force, pushing the equilibrium towards the desired product. This mechanism avoids the formation of stable metal-complex intermediates, thereby eliminating the risk of heavy metal contamination in the final API, a critical quality attribute for regulatory compliance. The robustness of this mechanism allows for a wide scope of substrates, as demonstrated by the successful synthesis of various derivatives bearing electron-donating and electron-withdrawing groups.
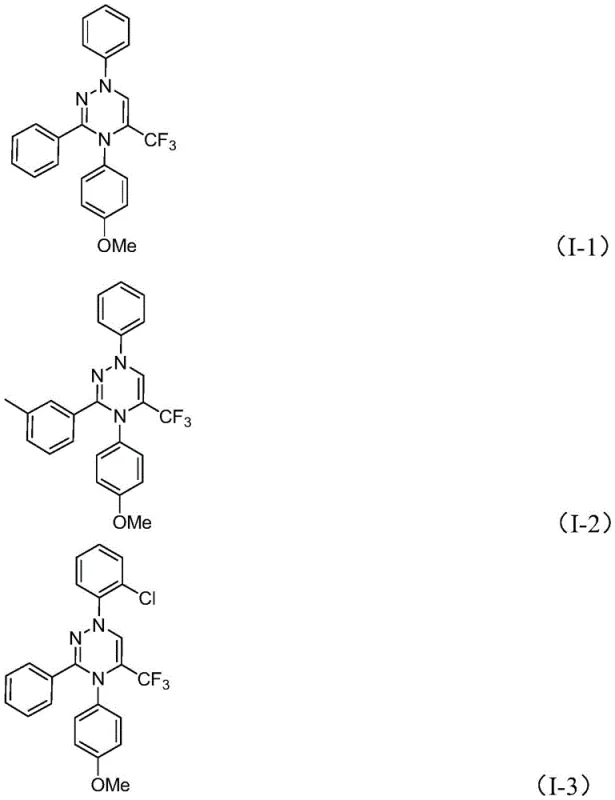
From an impurity control perspective, this mechanism offers distinct advantages over transition-metal catalyzed cycles. The absence of metals means there are no metal-ligand complexes to dissociate or residual catalysts to remove via scavenging resins, which simplifies the purification profile. The primary byproducts are inorganic salts and DMSO, both of which are easily removed during standard aqueous workup or chromatography. The reaction tolerates a broad range of functional groups on the aromatic rings, including halogens, alkoxy groups, and trifluoromethyl substituents, without significant side reactions. This high chemoselectivity ensures that the impurity spectrum remains clean and predictable, facilitating easier scale-up. The structural diversity achievable is exemplified by the synthesis of compounds I-1 through I-5, which feature varied substitution patterns on the phenyl and naphthyl rings, confirming the method's utility for generating diverse chemical libraries for drug screening.
How to Synthesize Trifluoromethyl 1,2,4-Triazines Efficiently
The practical execution of this synthesis is designed for maximum operational simplicity, making it ideal for both laboratory discovery and pilot plant operations. The protocol involves mixing the chlorohydrazone, trifluoroacetyl sulfur ylide, and potassium carbonate in a suitable organic solvent such as tetrahydrofuran (THF). The reaction mixture is then stirred at room temperature for a period of 10 to 14 hours, after which the completion of the reaction is monitored. Post-reaction processing is straightforward, typically involving filtration to remove inorganic salts followed by purification via column chromatography. This streamlined workflow minimizes unit operations and reduces the overall processing time compared to multi-step condensations. For detailed standardized synthesis procedures and specific molar ratios optimized for different substrates, please refer to the comprehensive guide below.
- Mix potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide in an organic solvent like THF.
- Stir the reaction mixture at room temperature (20-40°C) for 10-14 hours under an air atmosphere.
- Filter the mixture and purify the crude product via column chromatography to obtain the target triazine compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this technology translates into tangible strategic benefits regarding cost, reliability, and scalability. The elimination of expensive transition metal catalysts such as palladium or copper directly impacts the bill of materials, resulting in substantial cost savings per kilogram of produced intermediate. Furthermore, the ability to run the reaction under air at room temperature removes the capital expenditure associated with specialized inert atmosphere reactors and cryogenic cooling systems. This operational flexibility allows for the utilization of standard glass-lined or stainless steel reactors, enhancing the agility of the manufacturing facility to respond to market demands. The use of commodity chemicals like potassium carbonate and common solvents ensures a stable and resilient supply chain, mitigating risks associated with the sourcing of exotic reagents.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the removal of noble metal catalysts and the simplification of reaction conditions. By avoiding the need for rigorous exclusion of oxygen and moisture, energy consumption for heating, cooling, and nitrogen purging is drastically reduced. Additionally, the high atom economy of the [3+3] cycloaddition minimizes waste generation, lowering disposal costs. The straightforward workup procedure reduces labor hours and solvent usage during purification, contributing to a leaner manufacturing cost structure that enhances competitiveness in the global API market.
- Enhanced Supply Chain Reliability: The reliance on commercially available and stable starting materials such as acyl chlorides, hydrazines, and iodomethyl sulfoxide ensures a consistent supply flow. Unlike processes dependent on custom-synthesized building blocks with long lead times, the precursors for this method are widely sourced from multiple vendors. This redundancy in the supply base protects against disruptions and price volatility. Moreover, the robustness of the reaction conditions means that production schedules are less likely to be delayed by equipment failures related to complex environmental controls, ensuring on-time delivery for downstream customers.
- Scalability and Environmental Compliance: Scaling this process from gram to tonnage levels is facilitated by the exothermic nature of the reaction being manageable at room temperature, reducing safety risks associated with thermal runaways. The absence of heavy metals simplifies environmental compliance, as there is no need for extensive wastewater treatment to remove toxic metal residues. The generation of DMSO as a byproduct is manageable within standard solvent recovery systems. These factors collectively support the commercial scale-up of complex pharmaceutical intermediates while adhering to increasingly stringent green chemistry principles and regulatory standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aimed at clarifying the operational feasibility and strategic value for potential partners. Understanding these nuances is essential for R&D teams evaluating route selection and procurement teams assessing vendor capabilities.
Q: What are the primary advantages of this synthesis method over conventional condensation reactions?
A: Unlike traditional methods requiring harsh conditions or expensive catalysts, this patent describes a metal-free process operating at room temperature in air, significantly simplifying operational complexity and reducing raw material costs.
Q: Is the reaction sensitive to oxygen or moisture, requiring inert gas protection?
A: No, the reaction is remarkably robust and proceeds efficiently under an air atmosphere without the need for nitrogen protection or anhydrous conditions, which greatly enhances its suitability for large-scale manufacturing.
Q: What is the structural scope of the substrates tolerated in this reaction?
A: The method demonstrates excellent functional group tolerance, accommodating various substituted phenyl, naphthyl, and heteroaryl groups on the chlorohydrazone and sulfur ylide components, allowing for diverse library synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl 1,2,4-Triazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced heterocyclic intermediates play in accelerating drug development timelines. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless. We are committed to delivering high-purity trifluoromethyl 1,2,4-triazine derivatives that meet stringent purity specifications required by global regulatory bodies. Our rigorous QC labs employ state-of-the-art analytical techniques to verify identity and purity, guaranteeing that every batch supports your clinical and commercial success with consistency and reliability.
We invite you to collaborate with us to leverage this innovative synthesis technology for your next project. Our experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact our technical procurement team today to request specific COA data for our available inventory or to discuss route feasibility assessments for custom synthesis projects. Let us help you optimize your supply chain and reduce time-to-market for your vital therapeutic candidates.