Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Scalable Pharmaceutical Manufacturing
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Scalable Pharmaceutical Manufacturing
The strategic incorporation of trifluoromethyl groups into heterocyclic scaffolds represents a cornerstone of modern medicinal chemistry, significantly enhancing the electronegativity, metabolic stability, and lipophilicity of drug candidates. Patent CN111423381A introduces a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds, addressing the critical need for efficient synthetic routes in the development of high-purity pharmaceutical intermediates. This technology leverages a transition metal palladium-catalyzed carbonylation tandem reaction, utilizing inexpensive and commercially accessible starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salts. The significance of this innovation is underscored by the prevalence of imidazole motifs in bioactive molecules, ranging from antihistamines to complex natural products, as illustrated in the structural diversity of known pharmacophores.
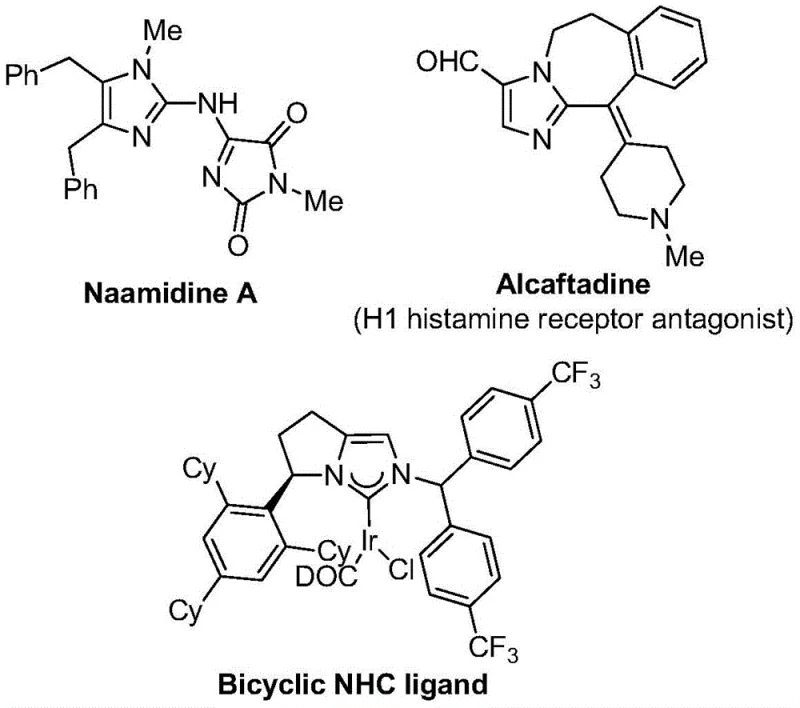
For R&D directors and process chemists, the ability to access these fluorinated heterocycles through a robust, one-pot transformation is invaluable. The patent details a protocol that not only simplifies the synthetic workflow but also ensures high reaction efficiency and broad substrate compatibility. By operating under mild thermal conditions, specifically at 30°C, this method mitigates the risks associated with thermal degradation of sensitive functional groups, thereby preserving the integrity of complex molecular architectures. This technical advancement positions the 2-trifluoromethyl imidazole scaffold as a more accessible building block for the next generation of therapeutic agents, facilitating rapid lead optimization and structure-activity relationship studies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nitrogen-containing heterocycles bearing trifluoromethyl functionalities has been fraught with challenges related to reagent availability, safety, and operational complexity. Conventional literature methods often rely on specialized trifluoromethyl synthons such as trifluorodiazoethane, which can be hazardous to handle and difficult to source on a commercial scale. Furthermore, existing protocols frequently necessitate harsh reaction conditions, including elevated temperatures or strong bases, which can limit the scope of compatible substrates and lead to the formation of undesirable byproducts. The reliance on such restrictive methodologies creates bottlenecks in the supply chain for high-purity pharmaceutical intermediates, increasing both the cost of goods and the lead time for process development. Additionally, the limited application of trifluoroethylimide acid halides in prior art has left a significant gap in the toolbox of synthetic organic chemists, restricting the diversity of accessible chemical space for drug discovery programs.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach disclosed in CN111423381A utilizes a sophisticated palladium-catalyzed carbonylation cascade that transforms simple, abundant precursors into valuable 2-trifluoromethyl imidazoles. This method employs trifluoroethylimidoyl chloride, a stable and readily available reagent, in conjunction with propargylamine and diaryliodonium salts to drive the reaction forward with remarkable efficiency. The core of this innovation lies in the seamless integration of carbon-nitrogen bond formation and carbonylation steps within a single reaction vessel, eliminating the need for intermediate isolation and purification. As depicted in the general reaction scheme, this tandem process allows for the modular assembly of the imidazole ring while simultaneously installing the critical trifluoromethyl group at the 2-position.
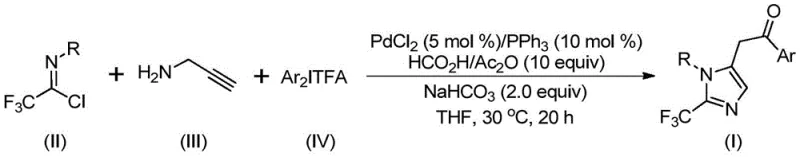
The operational simplicity of this new route is further enhanced by the use of a formic acid and acetic anhydride mixture as a convenient carbon monoxide surrogate, obviating the need for high-pressure CO gas cylinders which pose significant safety hazards in manufacturing environments. This shift towards safer, bench-stable reagents significantly lowers the barrier to entry for laboratories and production facilities alike. Moreover, the reaction proceeds effectively in common aprotic solvents such as tetrahydrofuran (THF), ensuring that the process is easily adaptable to existing infrastructure without requiring specialized equipment. The result is a versatile platform technology that enables the rapid synthesis of diverse 1,5-disubstituted imidazole derivatives, providing a powerful tool for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Pd-Catalyzed Carbonylation Tandem Reaction
A deep understanding of the catalytic cycle is essential for optimizing this process for commercial scale-up and ensuring consistent product quality. The proposed mechanism begins with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and propargylamine, generating a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization to facilitate the coordination of the alkyne moiety to the palladium center. The palladium catalyst, generated in situ from palladium chloride and triphenylphosphine, then mediates an aminopalladation of the alkyne, forming a key alkenyl palladium intermediate. This intermediate further isomerizes to a more stable alkyl palladium species, setting the stage for the critical carbonylation step. The insertion of carbon monoxide, released from the decomposition of the formic acid/acetic anhydride additive, yields an acyl palladium intermediate, effectively building the carbonyl functionality directly into the growing molecular framework.
Following carbonylation, the catalytic cycle advances through an oxidative addition step involving the diaryliodonium salt. This unique transformation generates a high-valent tetravalent palladium intermediate, a rare and highly reactive species that drives the final bond-forming event. The cycle concludes with a reductive elimination step, which releases the final 2-trifluoromethyl substituted imidazole product and regenerates the active palladium catalyst for subsequent turnover. From an impurity control perspective, this well-defined mechanistic pathway minimizes the formation of side products typically associated with radical-based trifluoromethylation methods. The use of specific ligands and additives helps to stabilize the palladium species throughout the cycle, preventing catalyst deactivation and ensuring high turnover numbers. This mechanistic clarity allows process chemists to fine-tune reaction parameters, such as the molar ratios of reagents and the choice of solvent, to maximize yield and purity, which is paramount for meeting the stringent specifications required for active pharmaceutical ingredients.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The practical implementation of this synthesis is designed for ease of execution, making it highly attractive for both laboratory-scale discovery and pilot-plant production. The standard operating procedure involves charging a reaction vessel with the palladium catalyst system, consisting of palladium chloride and triphenylphosphine, along with sodium bicarbonate as a base. To this mixture, the carbon monoxide source, comprising formic acid and acetic anhydride, is added alongside the organic solvent, preferably tetrahydrofuran, to ensure complete dissolution of all components. The key substrates—trifluoroethylimidoyl chloride, propargylamine, and the chosen diaryliodonium salt—are then introduced to the reaction mixture. The detailed standardized synthesis steps for optimizing yield and purity are outlined in the guide below.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, and a formic acid/acetic anhydride mixture in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 16 to 24 hours, followed by filtration and purification via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this patented methodology offers substantial strategic benefits regarding cost efficiency and supply continuity. The primary advantage stems from the utilization of starting materials that are not only commercially available but also economically priced compared to exotic fluorinating agents. Trifluoroethylimidoyl chloride can be synthesized rapidly from aromatic amines, which are abundant in the global chemical market, ensuring a stable and resilient supply chain. This reliance on commodity chemicals drastically reduces the risk of raw material shortages that often plague specialty synthesis routes. Furthermore, the mild reaction conditions eliminate the need for energy-intensive heating or cooling systems, leading to significant operational expenditure savings over the lifecycle of the product. The simplified workup procedure, involving basic filtration and silica gel chromatography, reduces the consumption of solvents and consumables, contributing to a leaner and more sustainable manufacturing process.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the elimination of expensive transition metal catalysts often required in cross-coupling reactions, as the palladium loading is kept low (5 mol%) while maintaining high efficiency. By avoiding the use of high-pressure carbon monoxide gas, the facility does not need to invest in specialized pressure-rated reactors or extensive safety monitoring systems, resulting in lower capital expenditure. The high atom economy of the tandem reaction ensures that a greater proportion of the raw material mass is converted into the desired product, minimizing waste disposal costs. Additionally, the ability to tolerate a wide range of functional groups means that protecting group strategies can often be avoided, shortening the overall synthetic sequence and reducing the total cost of goods sold for the final API intermediate.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route translates directly into improved supply chain reliability for downstream customers. Since the reaction tolerates various substituents on the aryl rings, including electron-withdrawing and electron-donating groups, a single platform can be used to produce a diverse library of analogues without changing the core process parameters. This flexibility allows manufacturers to respond quickly to changing market demands or clinical trial requirements. The use of stable reagents like diaryliodonium salts, which can be prepared from aryl boronic acids and aryl iodides, ensures that the supply chain is not dependent on a single source of fragile intermediates. Consequently, lead times for high-purity pharmaceutical intermediates can be significantly reduced, enabling faster time-to-market for new drug candidates.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram levels is facilitated by the homogeneous nature of the reaction and the absence of hazardous gaseous reagents. The patent explicitly notes the potential for industrial large-scale production, indicating that heat transfer and mixing issues are minimal under the prescribed conditions. From an environmental standpoint, the generation of waste is minimized through the efficient use of reagents and the avoidance of toxic byproducts associated with older trifluoromethylation methods. The use of THF as a solvent allows for established recycling protocols, further aligning the process with green chemistry principles and regulatory compliance standards. This scalability ensures that the technology can support the transition from clinical supply to commercial manufacturing without the need for extensive process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this 2-trifluoromethyl imidazole synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear picture of the method's capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What are the key advantages of this palladium-catalyzed method over traditional trifluoromethylation?
A: This method utilizes cheap and readily available starting materials like trifluoroethylimidoyl chloride and operates under mild conditions (30°C), avoiding the harsh reagents often required in conventional trifluoromethyl syntheses.
Q: What is the substrate compatibility for this 2-trifluoromethyl imidazole synthesis?
A: The process demonstrates excellent functional group tolerance, accommodating various substituents on the aryl rings including methyl, tert-butyl, halogens, trifluoromethyl, and nitro groups at ortho, meta, or para positions.
Q: Is this synthesis suitable for industrial scale-up?
A: Yes, the patent explicitly states that the method can be expanded to the gram level and potentially for industrial large-scale production due to its simple operation and high reaction efficiency.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed carbonylation technology in accelerating the development of fluorinated pharmaceuticals. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the bench to the plant. Our state-of-the-art facilities are equipped to handle the specific requirements of this synthesis, including the safe management of palladium catalysts and the precise control of reaction parameters necessary to achieve stringent purity specifications. Our rigorous QC labs employ advanced analytical techniques to verify the identity and purity of every batch, guaranteeing that the 2-trifluoromethyl imidazole intermediates we supply meet the highest industry standards for safety and efficacy.
We invite you to collaborate with our technical procurement team to explore how this innovative synthesis can optimize your supply chain and reduce costs. By requesting a Customized Cost-Saving Analysis, you can gain a detailed understanding of the economic benefits specific to your target molecule. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project needs. Let us leverage our expertise in fine chemical intermediates to support your mission of bringing life-saving medicines to patients faster and more efficiently.