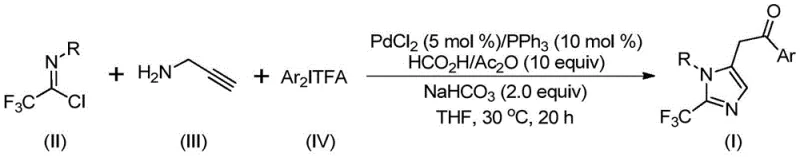
Advanced Pd-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Scalable Pharma Manufacturing
Advanced Pd-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Scalable Pharma Manufacturing
The rapid evolution of medicinal chemistry has placed a premium on fluorinated heterocycles, particularly those containing the trifluoromethyl moiety, due to their profound impact on the pharmacokinetic profiles of drug candidates. Patent CN111423381B introduces a groundbreaking methodology for the preparation of 2-trifluoromethyl substituted imidazole compounds, addressing a critical gap in the efficient construction of these privileged scaffolds. This technology leverages a sophisticated palladium-catalyzed multicomponent coupling strategy that merges trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts in a single operational step. For R&D directors and process chemists, this represents a significant leap forward, offering a pathway to complex nitrogen-containing heterocycles that are otherwise difficult to access with high regioselectivity and yield. The ability to introduce the trifluoromethyl group directly during the ring-forming event streamlines the synthetic route, eliminating the need for late-stage fluorination which often suffers from harsh conditions and poor functional group tolerance.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted imidazoles has been plagued by significant operational hazards and synthetic inefficiencies that hinder large-scale production. Traditional routes often rely on the use of trifluorodiazoethane, a highly explosive and unstable reagent that requires specialized handling equipment and poses severe safety risks in a manufacturing environment. Furthermore, existing methodologies frequently necessitate high temperatures, strong bases, or stoichiometric amounts of expensive metal oxidants, leading to elevated production costs and complex waste streams. The lack of modularity in older processes means that changing the substitution pattern on the imidazole ring often requires a complete redesign of the synthetic route, limiting the speed at which medicinal chemists can explore structure-activity relationships. These bottlenecks create substantial friction in the supply chain, resulting in long lead times and inconsistent quality for key pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology disclosed in CN111423381B utilizes a transition metal-catalyzed carbonylation cascade that operates under exceptionally mild conditions, typically at 30°C, thereby drastically reducing energy consumption and thermal stress on sensitive functional groups. By employing trifluoroethylimidoyl chloride and propargylamine as stable, commercially available building blocks, the process circumvents the need for hazardous diazo compounds entirely. The reaction utilizes a formic acid and acetic anhydride mixture as a safe, in-situ source of carbon monoxide, replacing the need for high-pressure CO gas cylinders and enhancing overall plant safety. This approach not only simplifies the operational procedure but also exhibits remarkable substrate versatility, allowing for the introduction of diverse aryl groups via diaryl iodonium salts without compromising reaction efficiency.
Mechanistic Insights into Palladium-Catalyzed Carbonylative Cyclization
The mechanistic elegance of this transformation lies in its intricate catalytic cycle, which orchestrates multiple bond-forming events with high precision. The reaction initiates with the formation of a trifluoroacetamidine intermediate through an intermolecular carbon-nitrogen bond promotion facilitated by the base. Following isomerization, the palladium catalyst engages in a palladation of the alkyne moiety of the propargylamine derivative, generating a crucial alkenyl palladium species. This intermediate undergoes further isomerization to an alkyl palladium complex, setting the stage for the carbonylation step where the in-situ generated carbon monoxide inserts into the palladium-carbon bond to form an acyl palladium intermediate. The cycle culminates in the oxidative addition of the diaryl iodonium salt to generate a transient tetravalent palladium species, followed by reductive elimination to forge the final carbon-carbon bond and release the 2-trifluoromethyl imidazole product while regenerating the active catalyst.
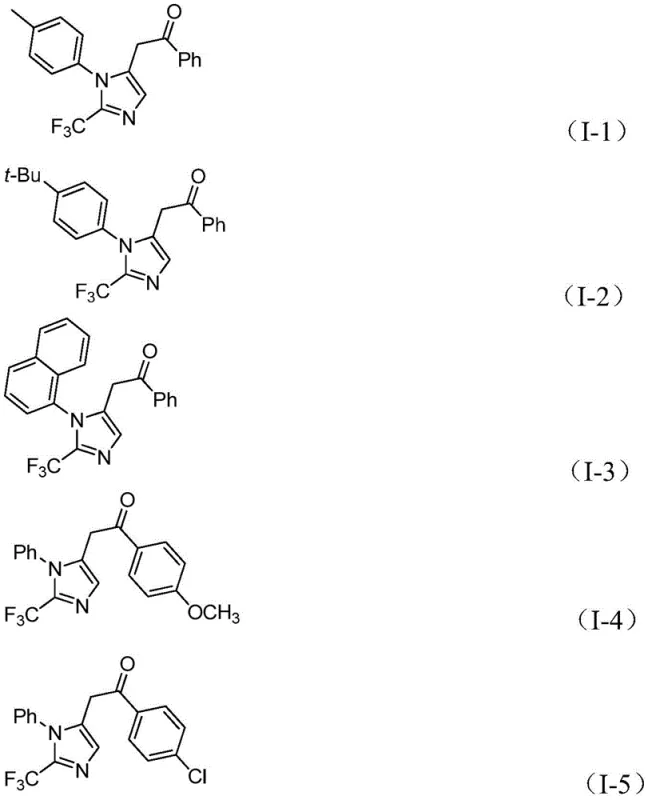
From an impurity control perspective, this mechanism offers distinct advantages by minimizing side reactions commonly associated with radical pathways or uncontrolled polymerization of alkynes. The use of diaryl iodonium salts as electrophiles ensures high chemoselectivity, as these reagents are less prone to homocoupling compared to traditional aryl halides under these specific conditions. Furthermore, the mild reaction temperature of 30°C suppresses thermal decomposition of the trifluoromethyl group and prevents the formation of tar-like byproducts that often complicate downstream purification. The robustness of the catalytic system allows for the tolerance of various substituents, including electron-withdrawing nitro and halogen groups, as well as electron-donating methoxy and alkyl groups, ensuring a clean reaction profile across a broad range of substrates.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The practical implementation of this synthesis is designed for ease of execution in both laboratory and pilot plant settings, requiring standard glassware and readily available reagents. The protocol involves a straightforward one-pot procedure where all components are mixed in an aprotic solvent like tetrahydrofuran, eliminating the need for intermediate isolation or inert atmosphere techniques beyond standard Schlenk line practices. The reaction proceeds to completion within 16 to 24 hours, after which simple filtration and silica gel chromatography yield the pure product. For detailed operational parameters and specific molar ratios optimized for different substrates, please refer to the standardized synthesis guide below.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, and formic acid in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 16 to 24 hours, then filter and purify via column chromatography to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented technology translates into tangible strategic benefits that extend far beyond simple yield improvements. The shift towards safer, more stable starting materials fundamentally alters the risk profile of the manufacturing process, reducing insurance liabilities and regulatory burdens associated with hazardous reagents. By eliminating the need for high-pressure reactors or cryogenic conditions, capital expenditure requirements for new production lines are significantly lowered, allowing for faster deployment of manufacturing capacity. The streamlined workflow reduces the number of unit operations, which directly correlates to lower labor costs and reduced solvent consumption, aligning perfectly with modern green chemistry initiatives and sustainability goals.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of inexpensive catalyst systems, specifically palladium chloride and triphenylphosphine, which are vastly more affordable than specialized ligand-catalyst complexes often required in cross-coupling reactions. The avoidance of exotic fluorinating agents and the use of commodity chemicals like formic acid and acetic anhydride as the carbonyl source further drive down the raw material cost per kilogram. Additionally, the high atom economy of the multicomponent reaction ensures that a greater proportion of the input mass is converted into the desired product, minimizing waste disposal costs and maximizing overall process efficiency.
- Enhanced Supply Chain Reliability: Sourcing stability is a critical concern for long-term projects, and this method relies on building blocks that are widely available from multiple global suppliers, mitigating the risk of single-source dependency. The robustness of the reaction conditions means that production is less susceptible to disruptions caused by minor fluctuations in utility supplies, such as cooling water temperature variations, ensuring consistent batch-to-batch quality. The scalability of the process from gram to multi-kilogram levels without significant re-optimization allows for seamless transition from clinical trial material supply to commercial launch volumes, securing the continuity of supply for downstream API manufacturers.
- Scalability and Environmental Compliance: The environmental footprint of this synthesis is markedly lower than conventional alternatives, primarily due to the absence of toxic carbon monoxide gas handling and the reduction in hazardous waste generation. The mild operating temperature reduces the energy load on the facility, contributing to a lower carbon footprint for the manufactured intermediates. Furthermore, the simplified workup procedure, which avoids complex aqueous quenches or extensive extraction sequences, facilitates easier solvent recovery and recycling, supporting the implementation of circular economy principles within the chemical manufacturing site.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific projects, we have compiled answers to common inquiries regarding the reaction parameters and scope. These insights are derived directly from the experimental data and optimization studies detailed in the patent literature, providing a realistic expectation of performance. Understanding these nuances is essential for effective process development and risk assessment prior to technology transfer.
Q: What are the critical reaction conditions for this synthesis?
A: The reaction operates under remarkably mild conditions, specifically at 30°C for 16 to 24 hours, utilizing THF as the preferred solvent and a PdCl2/PPh3 catalyst system.
Q: What is the source of the carbonyl group in this transformation?
A: Instead of using toxic carbon monoxide gas, the process employs a safe in-situ generation system using formic acid and acetic anhydride as the carbonyl source.
Q: Does this method tolerate diverse functional groups on the substrates?
A: Yes, the protocol demonstrates excellent substrate compatibility, successfully accommodating electron-donating groups like methoxy and tert-butyl, as well as electron-withdrawing groups like nitro and halogens.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced heterocyclic intermediates play in the development of next-generation therapeutics and functional materials. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to industrial manufacturing is seamless and efficient. We are committed to delivering high-purity 2-trifluoromethyl imidazole derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify identity and assay.
We invite you to collaborate with us to leverage this cutting-edge synthesis technology for your upcoming projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality standards. Please contact us today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary targets, and let us help you accelerate your path to market with confidence.