Advanced Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Commercial API Production
The pharmaceutical and agrochemical industries continuously seek robust synthetic routes for nitrogen-containing heterocycles due to their prevalence in bioactive molecules. Patent CN111978265B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole derivatives, a structural motif found in blockbuster drugs such as Maraviroc, Triazolam, Sitagliptin, and Deferasirox. The introduction of a trifluoromethyl group into these heterocyclic frameworks significantly enhances electronegativity, metabolic stability, and lipophilicity, making these derivatives highly desirable for drug discovery programs. This new methodology represents a significant leap forward in process chemistry, offering a streamlined approach that bypasses the limitations of classical synthesis.
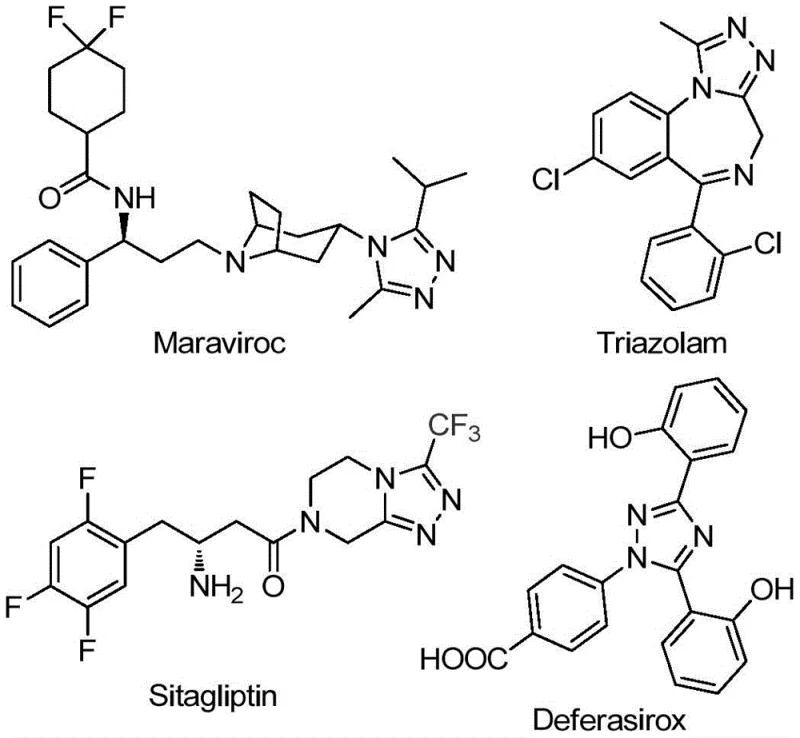
As a reliable pharmaceutical intermediate supplier, understanding the nuances of such patented technologies is crucial for ensuring supply chain continuity. The disclosed method utilizes a tandem cyclization strategy promoted by ferric chloride, enabling the efficient construction of the triazole core under relatively mild conditions. This innovation not only simplifies the manufacturing process but also opens up new avenues for designing diverse libraries of trifluoromethylated heterocycles, thereby accelerating the development of next-generation therapeutics and functional materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted 1,2,4-triazoles has been plagued by significant operational challenges that hinder cost reduction in API manufacturing. Traditional literature methods often rely on the condensation of 3,5-ditrifluoromethyl-1,3,4-oxadiazoles with primary amines or the cyclization of trifluoromethyl hydrazides with amidines. These conventional pathways frequently demand harsh reaction conditions, including extreme temperatures or the use of hazardous reagents, which complicate safety protocols and waste management. Furthermore, many existing methods suffer from narrow substrate scopes, particularly failing to accommodate alkyl hydrazones, which severely limits the chemical diversity accessible to medicinal chemists. The low reaction yields and lengthy multi-step sequences associated with these older techniques result in poor atom economy and inflated production costs, making them less attractive for large-scale commercial applications.
The Novel Approach
In stark contrast, the novel approach detailed in patent CN111978265B utilizes a simple yet highly effective strategy starting from cheap and readily available acyl hydrazides and trifluoroethylimidoyl chlorides. This method employs ferric chloride as a promoter, facilitating a smooth cyclization that tolerates a wide range of functional groups without the need for stringent anhydrous or oxygen-free environments. By operating under milder thermal conditions and utilizing common organic solvents like 1,4-dioxane, this process drastically simplifies the operational workflow. The ability to successfully synthesize 3-alkyl substituted 1,2,4-triazoles, a feat unachievable with previous tandem cyclization methods, underscores the versatility of this new route. This breakthrough provides a practical solution for the high-purity production of complex heterocycles, directly addressing the pain points of scalability and efficiency in modern chemical synthesis.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The mechanistic pathway of this transformation involves a sophisticated interplay between base-promoted nucleophilic attack and Lewis acid-catalyzed dehydration. Initially, sodium bicarbonate facilitates the intermolecular formation of a carbon-nitrogen bond between the trifluoroethylimidoyl chloride and the hydrazide, generating a trifluoroacetamidine intermediate. This step is critical for setting up the molecular architecture required for ring closure. Subsequently, the addition of ferric chloride acts as a potent Lewis acid, activating the intermediate for an intramolecular dehydration condensation reaction. This dual-catalyst system ensures that the cyclization proceeds efficiently to form the final 5-trifluoromethyl substituted 1,2,4-triazole derivative with high regioselectivity.
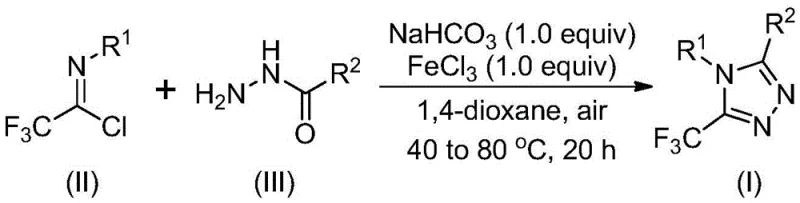
From an impurity control perspective, the mild nature of this catalytic system is paramount. Harsh acidic or basic conditions often lead to the decomposition of sensitive trifluoromethyl groups or the formation of polymeric byproducts. By carefully tuning the reaction temperature and employing a stepwise addition of reagents, the process minimizes side reactions, resulting in a cleaner crude product profile. The use of ferric chloride, a non-precious metal catalyst, also alleviates concerns regarding heavy metal contamination in the final active pharmaceutical ingredient, simplifying the downstream purification process. This mechanistic elegance translates directly into higher purity specifications and reduced processing time, which are key metrics for any commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Derivatives Efficiently
The synthesis protocol outlined in the patent offers a straightforward procedure adaptable for both laboratory research and pilot plant operations. The process begins with the dissolution of sodium bicarbonate, trifluoroethylimidoyl chloride, and the corresponding hydrazide in an aprotic organic solvent. The mixture is stirred at a moderate temperature to allow for the initial coupling, followed by the addition of the iron catalyst to drive the cyclization to completion. Detailed standardized synthesis steps for implementing this route are provided in the guide below, ensuring reproducibility and safety during execution.
- Mix sodium bicarbonate, trifluoroethylimidoyl chloride, and hydrazide in an aprotic organic solvent such as 1,4-dioxane.
- Stir the mixture at a moderate temperature range of 30 to 50 degrees Celsius for 8 to 16 hours to facilitate initial bond formation.
- Add ferric chloride catalyst and increase temperature to 70 to 90 degrees Celsius for 6 to 10 hours to complete the cyclization and dehydration.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers substantial strategic benefits beyond mere chemical efficiency. The shift towards this methodology addresses critical bottlenecks in the sourcing of key building blocks and the overall cost structure of production. By leveraging widely available commodity chemicals and eliminating the need for specialized reaction infrastructure, manufacturers can achieve significant operational improvements.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of inexpensive and abundant starting materials. Trifluoroethylimidoyl chlorides and acyl hydrazides are commercially accessible at competitive price points, unlike some exotic reagents required by alternative methods. Furthermore, the utilization of ferric chloride as a catalyst represents a drastic cost saving compared to precious metal catalysts often used in cross-coupling reactions. The elimination of rigorous anhydrous conditions reduces energy consumption associated with solvent drying and inert gas purging, leading to lower utility costs per kilogram of product produced.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the broad availability of the requisite raw materials. Since the starting materials are common industrial chemicals rather than custom-synthesized specialty items, the risk of supply disruption is minimized. The robustness of the reaction conditions means that production is less susceptible to delays caused by equipment failures related to high-pressure or high-temperature requirements. This reliability ensures consistent lead times for high-purity pharmaceutical intermediates, allowing downstream drug manufacturers to maintain stable inventory levels and meet market demand without interruption.
- Scalability and Environmental Compliance: The simplicity of the workup procedure, involving basic filtration and standard chromatography, facilitates easy scale-up from gram to multi-kilogram quantities. The process generates fewer hazardous byproducts compared to traditional methods that might utilize strong acids or toxic heavy metals. This aligns with increasingly stringent environmental regulations and corporate sustainability goals. The ability to run the reaction in air without specialized glovebox equipment further simplifies the engineering controls required for commercial scale-up, reducing capital expenditure on reactor modifications and safety systems.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for technical teams evaluating this route for potential integration into their manufacturing pipelines.
Q: What are the primary advantages of this FeCl3-catalyzed method over traditional triazole synthesis?
A: This method eliminates the need for harsh anhydrous or oxygen-free conditions required by older protocols. It utilizes inexpensive, commercially available starting materials like hydrazides and achieves high yields with a broad substrate scope, including alkyl hydrazones which were previously unreactive.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process is highly scalable. It employs simple workup procedures involving filtration and standard column chromatography. The use of cheap catalysts like ferric chloride and common solvents like 1,4-dioxane makes it economically viable for commercial scale-up.
Q: What types of substituents are tolerated in this triazole synthesis?
A: The reaction demonstrates excellent functional group tolerance. It accommodates various substituted aryl groups on both the imidoyl chloride and the hydrazide components, including methyl, methoxy, halogen, and trifluoromethyl groups at ortho, meta, or para positions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic methodologies like the FeCl3-promoted cyclization described in patent CN111978265B. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory processes are seamlessly translated into robust industrial realities. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole derivatives meets the exacting standards required for global pharmaceutical applications.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us to request specific COA data and comprehensive route feasibility assessments, and let us demonstrate how our expertise can optimize your supply chain for high-purity pharmaceutical intermediates.