Advanced Manufacturing of Cinepazide Maleate: A Cost-Effective Route for Cardiovascular API Intermediates
Introduction to Patent CN101260092B and Strategic Process Innovation
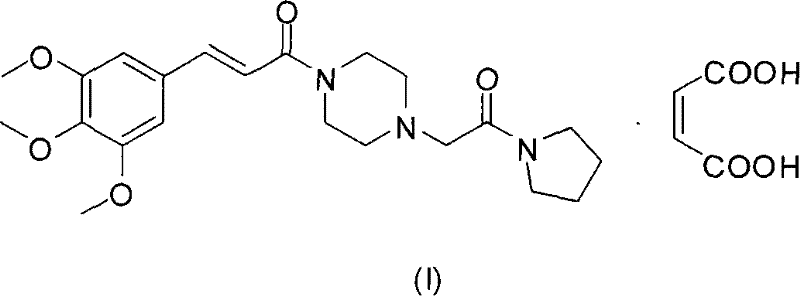
The pharmaceutical landscape for cerebrovascular therapeutics demands intermediates of exceptional purity and consistent polymorphic stability, a challenge effectively addressed by the methodology disclosed in Chinese Patent CN101260092B. This intellectual property outlines a robust, scalable synthesis for Cinepazide Maleate, a critical piperazine-derivative active pharmaceutical ingredient known for enhancing brain metabolism and stabilizing cell membranes against ischemic reperfusion injury. Unlike legacy processes that rely on hazardous volatile organic compounds, this novel approach leverages aqueous media for the initial acylation and acetate esters for the coupling reaction, fundamentally shifting the safety and economic profile of production. By integrating precise pH control mechanisms and a specialized mixed-solvent crystallization technique, the patent ensures the exclusive formation of the stable crystal form with a melting point of 173~174℃, thereby eliminating batch-to-batch variability that often plagues generic manufacturing. For global supply chain stakeholders, this represents a pivotal opportunity to secure a reliable pharmaceutical intermediates supplier capable of delivering high-specification materials without the environmental liabilities of traditional chlorinated solvent systems.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
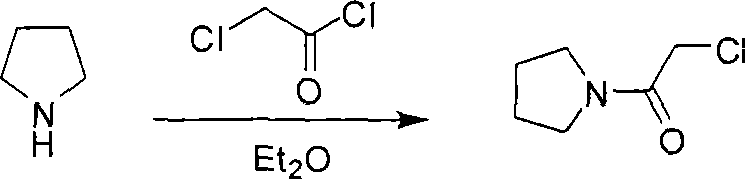
Historical synthetic routes for Cinepazide Maleate and its precursors have been fraught with significant operational hazards and purification bottlenecks that hinder industrial scalability. Prior art, such as the methods referenced in Farmaco (1963) and early patents, frequently utilized diethyl ether as a solvent for the synthesis of chloroacetyl pyrrolidine, a practice that introduces severe explosion risks due to ether's low boiling point and high flammability during exothermic acylation. Furthermore, alternative pathways described in US3634411 relied on anhydrous benzene, a known carcinogen with strict regulatory limits, creating substantial waste disposal costs and worker safety liabilities for modern facilities. The purification of the key intermediate, 1-piperazine acetylpyrrolidine, was traditionally complicated by the use of excessive piperazine and difficult distillation steps, often resulting in persistent di-substituted byproducts that are chemically similar and notoriously difficult to separate via standard chromatography. These legacy inefficiencies not only inflate the cost of goods sold but also introduce unpredictable variables into the supply chain, making it difficult for procurement managers to guarantee consistent quality for downstream API synthesis.
The Novel Approach
The methodology presented in CN101260092B decisively breaks away from these archaic constraints by introducing a water-based acylation protocol that operates safely at ambient temperatures between 20~30℃. By reacting chloroacetylpyrrolidine with piperazine hydrochloride in an aqueous environment, the process inherently suppresses the formation of di-substituted impurities through controlled protonation states, allowing for the selective isolation of the mono-acylated product simply by adjusting the pH to the alkaline range of 10~13. Subsequent coupling with 3,4,5-trimethoxycinnamoyl chloride is performed in ethyl acetate, a greener solvent that offers superior recovery rates compared to methylene dichloride, significantly reducing solvent loss and environmental impact. This strategic substitution of reaction media not only simplifies the workup procedure—eliminating the need for complex vacuum distillations—but also enhances the overall yield, with embodiment data demonstrating isolated yields of approximately 90% for the intermediate and 80% for the free base. Such improvements translate directly into a more resilient manufacturing process that is easier to control and validate under GMP conditions.
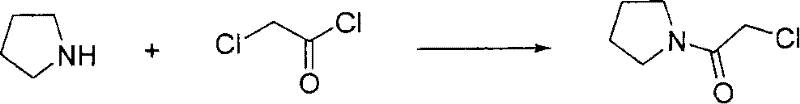
Mechanistic Insights into Aqueous Acylation and Polymorphic Control
The core chemical innovation of this patent lies in the sophisticated manipulation of acid-base equilibria to drive selectivity during the formation of the piperazine intermediate. In the aqueous phase, piperazine exists in equilibrium between its free base and hydrochloride salt forms; by carefully maintaining the reaction pH within the narrow window of 10 to 13 using alkaline agents like NaOH or K2CO3, the process ensures that only one nitrogen atom of the piperazine ring is sufficiently nucleophilic to attack the chloroacetyl group. This mechanistic precision prevents the second acylation event that typically leads to bis-acylated impurities, which are common in non-aqueous systems where base strength is harder to modulate. Following the reaction, the product is extracted into chloroform, leaving the unreacted piperazine salts in the aqueous layer, a partitioning strategy that serves as an initial bulk purification step before the final wet distillation removes trace amines. This multi-stage purification logic ensures that the subsequent coupling reaction proceeds with high efficiency, as the presence of free amine impurities would otherwise consume the valuable cinnamoyl chloride reagent and generate hard-to-remove urea-like side products.
Beyond reaction selectivity, the patent provides critical insights into the thermodynamics of crystallization required to secure the bioactive polymorph of Cinepazide Maleate. The inventors discovered that recrystallization from single solvents like ethanol or acetone alone fails to produce the stable crystal lattice necessary for long-term shelf life and consistent dissolution profiles. Instead, the use of a specific binary solvent system comprising alcohols (ethanol or isopropanol) and acetone in a volume ratio ranging from 1:3 to 1:4 creates a unique solubility gradient during reflux. This mixed solvent environment facilitates the orderly assembly of the maleate salt molecules into the thermodynamically stable form, characterized by a sharp melting point of 173~174℃ and a DSC endotherm at 182.6℃. Understanding this solvate-mediated transformation is vital for R&D directors, as it dictates that any deviation in solvent ratios or cooling rates could result in metastable forms that might convert over time, potentially altering the drug's bioavailability and regulatory compliance status.
How to Synthesize Cinepazide Maleate Efficiently
The synthesis of this high-value cardiovascular intermediate follows a logical, four-stage progression designed to maximize yield while minimizing hazardous waste generation. The process begins with the preparation of the acylating agent, followed by the crucial aqueous coupling with piperazine, then the final condensation with the trimethoxycinnamoyl moiety, and concludes with a rigorous salt formation and crystallization protocol. Each step has been optimized to operate under mild conditions, avoiding extreme temperatures or pressures that would require specialized reactor hardware. The detailed standardized operating procedures, including exact reagent stoichiometry, agitation speeds, and filtration parameters, are essential for replicating the high purity (99.65%) reported in the patent embodiments. For technical teams looking to implement this route, adherence to the specific pH adjustments and solvent mixing ratios described below is paramount to achieving the desired crystal habit and impurity profile.
- React chloroacetylpyrrolidine with piperazine hydrochloride in water, adjusting pH to 10-13 to isolate 1-piperazine acetylpyrrolidine.
- Couple the intermediate with 3,4,5-trimethoxycinnamoyl chloride in ethyl acetate at room temperature to form the free base.
- Form the maleate salt and recrystallize using a specific ethanol-acetone mixture to obtain the stable crystal form (MP 173-174°C).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of the CN101260092B synthesis route offers profound advantages in cost reduction in pharmaceutical intermediates manufacturing by fundamentally altering the input cost structure. The replacement of expensive, anhydrous organic solvents like ether and benzene with commodity-grade water and recoverable ethyl acetate drastically lowers raw material expenditures and reduces the energy burden associated with solvent drying and recovery. Furthermore, the elimination of complex vacuum distillation steps for intermediate purification shortens the cycle time per batch, allowing for higher throughput in existing reactor trains without the need for capital-intensive equipment upgrades. This streamlined workflow not only reduces direct labor costs but also minimizes the risk of batch failures due to operator error in handling hazardous volatiles, thereby protecting the overall asset utilization rate of the manufacturing facility.
- Cost Reduction in Manufacturing: The transition to an aqueous-first strategy eliminates the need for costly drying agents and inert atmosphere protections typically required for moisture-sensitive acylations in organic solvents. By utilizing water as the primary reaction medium for the piperazine coupling, the process avoids the purchase and disposal of large volumes of chlorinated solvents, which are increasingly subject to stringent environmental taxes and disposal fees. Additionally, the high recovery rate of ethyl acetate in the second step means that solvent make-up costs are negligible, contributing to a significantly lower variable cost per kilogram of finished product compared to legacy benzene-based routes.
- Enhanced Supply Chain Reliability: The reliance on widely available, non-proprietary reagents such as piperazine hydrochloride, sodium hydroxide, and common esters ensures that the supply chain is not vulnerable to shortages of exotic catalysts or specialized reagents. The robustness of the reaction conditions, which tolerate ambient temperatures and do not require cryogenic cooling, further enhances reliability by reducing dependence on complex utility systems like chillers or steam jets that can be points of failure in older plants. This operational simplicity allows for flexible production scheduling and rapid scale-up from pilot to commercial quantities, ensuring that procurement managers can secure consistent volumes of high-purity pharmaceutical intermediates even during periods of market volatility.
- Scalability and Environmental Compliance: The process design inherently aligns with green chemistry principles by minimizing the E-factor (mass of waste per mass of product) through the use of water and recyclable esters. The absence of carcinogenic benzene and highly flammable ether removes significant regulatory hurdles related to worker exposure limits and explosion-proof zoning, facilitating easier permitting for capacity expansion in regulated jurisdictions. Moreover, the high purity of the crude product achieved through pH-controlled extraction reduces the load on downstream purification units, resulting in less solvent waste generated during final recrystallization and a smaller environmental footprint for the entire manufacturing lifecycle.
Frequently Asked Questions (FAQ)
The following technical inquiries address common concerns regarding the implementation and quality assurance of this specific synthesis pathway. These answers are derived directly from the experimental data and comparative analysis provided within the patent specification, offering clarity on critical process parameters. Understanding these nuances is essential for quality assurance teams and process engineers who are evaluating the feasibility of technology transfer or vendor qualification for this specific intermediate.
Q: Why is water preferred over organic solvents for the initial acylation step?
A: Using water eliminates the explosion risks associated with ether and reduces toxicity compared to benzene or methylene chloride, while simplifying pH-controlled isolation of the mono-substituted product.
Q: How does the new method ensure the stability of the crystal form?
A: The process utilizes a specific mixed solvent system of alcohols and acetone during reflux, which guarantees the formation of the thermodynamically stable polymorph with a melting point of 173-174°C.
Q: What are the purity levels achievable with this synthesis route?
A: The optimized protocol, including activated carbon decolorization and precise pH adjustments, consistently yields cinepazide free base with purity exceeding 99.65% by HPLC.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cinepazide Maleate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of cerebrovascular therapeutics hinges on the availability of intermediates with uncompromising quality and supply security. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate pH controls and mixed-solvent crystallization techniques described in CN101260092B are executed with precision at every scale. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the critical melting point range of 173~174℃ that defines the stable polymorph, guaranteeing that every batch meets the exacting standards required for global regulatory filings.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this cost-effective and environmentally sustainable manufacturing route. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the potential reductions in solvent and waste disposal costs specific to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your supply chain for high-purity pharmaceutical intermediates is built on a foundation of scientific excellence and commercial reliability.